Research Paper
Senolytics Reduce Endothelial Cell DNA Damage and Telomere Dysfunction Despite Reductions in Telomere Length
Authors
Samuel I. Bloom,1 Eric Tuday,2,3 Md Torikul Islam,1 Venkateswara R. Gogulamudi,4 Lisa A. Lesniewski,1,3,4,5 and Anthony J. Donato1,3,4,5,6,*
1Department of Nutrition and Integrative Physiology, The University of Utah, Salt Lake City, UT, USA
2Department of Internal Medicine, Division of Cardiology, The University of Utah, Salt Lake City, UT, USA
3Veteran’s Affairs Medical Center-Salt Lake City, Geriatric, Research, Education, and Clinical Center, Salt Lake City, UT, USA
4Department of Internal Medicine, Division of Geriatrics, The University of Utah, Salt Lake City, UT, USA
5Nora Eccles Harrison Cardiovascular Research and Training Institute, The University of Utah, Salt Lake City, UT, USA
6Department of Biochemistry, The University of Utah, Salt Lake City, UT, USA
*Corresponding author: tony.donato@hsc.utah.edu
DOI:https://doi.org/10.59368/agingbio.20230007
Abstract
Aging results in cellular damage that can induce cell cycle arrest known as cellular senescence. Endothelial cells are one of the first cell types to become senescent in advancing age and contribute to age-related cardiovascular diseases. Drugs known as senolytics reduce endothelial cell senescence in cell culture. From a translational perspective, a key question is whether this occurs in vivo and if remaining cells appear healthier and display fewer hallmarks of cellular aging. In this study, we treated old mice with the senolytic cocktail dasatinib and quercetin (D+Q) or a vehicle control. In 24-month-old mice, D+Q treatment reduced p21 gene expression in carotid artery endothelial cells, indicative of reductions in senescence. In lung endothelial cells, we examined DNA damage, telomere dysfunction (DNA damage signaling at telomeres), and telomere length, which are hallmarks of aging associated with senescence and other deleterious effects on cellular function. D+Q treatment resulted in fewer endothelial cells with DNA damage and dysfunctional telomeres. Surprisingly, D+Q reduced endothelial cell telomere length, yet this did not result in critically short telomeres and thus telomere dysfunction. Mice have longer telomeres than humans; therefore, future studies on the effect of senolytics on telomere length are warranted. Collectively, this study provides important evidence on the effect of senolytics, including that they clear senescent endothelial cells in vivo, which reduces DNA damage and telomere dysfunction. These data indicate that the clearing of senescent endothelial cells in old age leaves behind a population of cells that exhibit fewer hallmarks of vascular aging.
Introduction
Aging results in the accumulation of cellular damage that can activate tumor suppressor pathways leading to permanent cell cycle arrest known as cellular senescence1. Senescent cells are pro-oxidative and pro-inflammatory and thus contribute to a multitude of age-related diseases2. Accordingly, drugs known as senolytics that selectively induce cell death in senescent cells have tremendous efficacy to delay or reduce age-related diseases in preclinical studies and are currently under investigation in clinical trials for use to treat a host of human diseases3. Several senolytic drugs being investigated were originally identified based on their ability to induce cell death in senescent endothelial cells in cell culture4,5. However, there is a paucity of data demonstrating whether senolytic drugs reduce endothelial cell senescence in vivo. Furthermore, from a translational perspective, a key question is whether following senolysis, the remaining cells appear healthier and display fewer hallmarks of cellular aging.
The vascular endothelium is a monolayer of cells that lines the lumen of the vascular network and interacts with the circulating milieu, including damaging hemodynamic forces and pulsatile pressures, high partial pressures of oxygen, and circulating metabolites6. Through constant exposure to this unique hostile environment, endothelial cells are one of the first cell types to become senescent with advancing age6,7. Importantly, mounting evidence demonstrates that endothelial cell senescence contributes to the development of cardiovascular disease6,8, the leading cause of death9,10. The vascular endothelium has many important roles which are impaired following the induction of senescence including the formation of new blood vessels, regulation of the trafficking of immune cells and macromolecules between blood and peripheral tissues, and modulation of vascular tone and blood flow6. This understanding of endothelial cell senescence highlights the importance of investigations that provide insight into the impact of clearing senescent endothelial cells.
Although many cellular stressors can induce senescence, total DNA damage and damage at repeat sequences at the ends of chromosomes known as telomeres are hallmarks of aging that occur in endothelial cells with advancing age and are robust inducers of senescence6,8,11. Furthermore, the consequences of these aging hallmarks extend beyond senescence, because they can impair transcription, DNA replication, and lead to mutations12. In this study, we tested the hypothesis that chronic administration of a senolytic cocktail would reduce the burden of endothelial cell senescence and molecular hallmarks of aging including DNA damage and telomere dysfunction.
Methods
Animals
Animal studies were in compliance with the Guide and Use of Laboratory Animals and were approved by The University of Utah and Veteran’s Affairs Medical Center-Salt Lake City. Old male C57BL/6 mice were obtained from the National Institute on Aging colony maintained at Charles River Inc. Mice were housed at the Veteran’s Affairs Medical Center-Salt Lake City Animal Facility in standard shoebox cages on a 12:12 light–dark cycle with access to food and water ad libitum.
Senolytic and vehicle treatments
At 21 months of age, 10 mice received the senolytic drug cocktail dasatinib (D, 5 mg/kg body mass) and quercetin (Q, 50 mg/kg body mass) on three consecutive days every two weeks for three months via oral gavage as previously described13. 10 control mice were treated with vehicle control solution containing 10% polyethylene glycol 4000. Following treatment, mice were sacrificed at 24 months of age.
Lung endothelial cell isolation
Lungs from three animals from the same group were collected and placed into a cold isolation media containing Dulbecco’s Modified Eagle's Medium (DMEM, Sigma-Aldrich, D0819, 4500 mg/L glucose, stable glutamine, and sodium bicarbonate, without sodium pyruvate), 20% fetal bovine serum (FBS, Gibco, 26140095), and 1% penicillin/streptomycin. Lungs were then mechanically digested and placed into a dissociation media containing 2 mg/mL collagenase type II in DMEM and incubated at 37°C for 40 minutes. Lungs were further mechanically digested using a sterile 6″-long 14-gauge metal cannula and triturated into a single cell suspension, pipetted through a 70 μm disposable cell strainer, and washed with 10 mL of isolation media. Cell suspensions were spun at 1000 rpm × 10 minutes, supernatant was aspirated, and cells were resuspended in 2 mL 0.1% bovine serum albumin (BSA, Sigma-Alrdich A9647) in 1× phosphate buffered saline (PBS, Gibco, 10010023). Cells were then tumbled with anti-platelet endothelial cell adhesion molecule (PECAM) (CD-31) beads bound to sheep anti-rat IgG Dynabeads for 30 minutes, and cells were isolated using a magnetic separator. Cells were grown on glass coverslips until 60%–70% confluent at which time they were fixed in 4% paraformaldehyde (PFA) for 15 minutes at room temperature.
Immunofluorescence fluorescent in situ hybridization (IF-FISH)
IF-FISH was performed as previously described11. Briefly, cells were placed in cold 100% methanol at −20°C for 15 minutes, rehydrated in 1× PBS at room temperature for 5 minutes, and then placed in a blocking solution containing 1 mg/mL BSA, 3% goat serum, 0.1% triton X-100, 1 mM EDTA, all in 1× PBS for 30 minutes at room temperature. Cells were then incubated for 1 hour at room temperature in 1:500 53BP1 antibody (Novus Biologicals, NB 100-0304, Rabbit) in blocking solution. Samples were washed 3 × 5 minutes in 1× PBS and then incubated for 1 hour at room temperature in 1:500 Alexa Fluor 555 in blocking solution (Invitrogen, A-21429, Goat anti-Rabbit) in blocking solution. Cells were dehydrated in 70%, 95%, and 100% ethanol for 5 minutes each. Ethanol was aspirated, and samples were allowed to dry very briefly before hybridization solution containing 2% Tris HCl, 60% formamide, and 5% blocking reagent from a 10% Roche Stock (blocking reagent for nucleic acid hybridization, stock dissolved in maleic acid buffer containing 100 mM maleic acid, 150 mM NaCl, pH 7.4, Milipore Sigma), and 1:200 tel probe (Integrated DNA Technologies, 5Alex488N/CC CTA ACC CTA ACC CTA A, purification: high-performance liquid chromatography) all in deionized H2O (diH2O) was added. Samples were incubated at 60°C for 10 minutes and at 85°C for 10 minutes. Cells were then gently agitated in washing solution containing 2× saline sodium citrate (SSC) and 0.1% Tween 20 at 60°C for 10 minutes two times. Cells were then washed in 2× SSC, 1× SSC, and then diH2O all for 10 minutes at room temperature. Samples were allowed to dry very briefly and then mounted with 4′,6-diamidino-2-phenylindole (DAPI) Fluoromount G (VWR—Catalog #102092-102), and weighted with 1 kg weight for 5 minutes and stored in a dark container until imaging.
Imaging and analysis
Samples were imaged on an Olympus Fluoview FV1000 confocal microscope at 100× magnification. All samples were imaged using the same acquisition settings. 1 μm z-slices were taken through the entirety of each nucleus, and the same number of z-slices was stacked for each cell for analysis. Images were analyzed using the Telometer Plugin for ImageJ (https://demarzolab.pathology.jhmi.edu/telometer/index.html), and telomere length was determined from fluorescence intensity. Both total 53BP1 foci and 53BP1 foci that colocalized with the telomere signal (telomere dysfunction-induced foci [TIF]) were quantified. About 274 total cells from vehicle-treated mice and 260 total cells from D+Q-treated mice were analyzed, amounting to 91.0 ± 4.5 cells/experiment from three vehicle experiments, and 86.7 ± 6.9 cells/experiment from three D+Q experiments. In total, 13,686 individual telomeres were analyzed from vehicle cells and 11,243 from D+Q cells.
Carotid artery endothelial cell mRNA isolation and quantitative real-time polymerase chain reaction (qRT-PCR)
Carotid arteries were excised and cannulated onto a 30-gauge dispensing needle attached to a 1 mL syringe. 100 μL of Qiazol lysis reagent was then flushed through each carotid artery, with the endothelial-enriched effluent from the arteries of the same mice collected in a 1.7 mL Eppendorf tube, and then snap frozen in liquid nitrogen. Total RNA from the endothelial enriched effluent was isolated using the RNAeasy Mini Kit (Qiagen) according to the manufacturer’s protocol and converted to cDNA using the QuantiTect Reverse Transcription Kit (Qiagen) according to the manufacturer’s protocol. Quantitative real-time PCR was performed using SsoFast EvaGreen Supermixes (Bio-Rad) and Bio-Rad CFX™ Real Time system. The 2−ΔΔCt was used to quantify relative gene expression. Primer sequences were as follows: 18s—Forward, 5′-TAGAGGGACAAGTGGCGTTC-3′, Reverse, 5′-CGCTGAGCCAGTCAGTGT-3′; p21—Forward, 5′-CCTGGTGATGTCCGACCTG-3′, Reverse, 5′-CCATGAGCGCATCGCAATC-3′.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 9.4.0. Group differences were determined by two-tailed t tests or repeated measures two-way analysis of variance (ANOVA) with least significant difference (LSD) post hoc tests. To assess the relationship between mean telomere length and TIF, a simple linear regression analysis was performed. Frequency distributions of telomere length were analyzed using a Mann–Whitney test. Statistical significance was set at p < 0.05, and data are presented as mean ± standard error of the mean (SEM).
Results
Effect of senolytics on endothelial cell senescence and DNA damage
To determine if senolytic administration reduced endothelial cell senescence, we examined mRNA expression of the cyclin-dependent kinase inhibitor that enforces senescence, p21, in endothelial cell enriched carotid artery effluents. D+Q-treated mice displayed an ∼57% reduction in endothelial p21 gene expression compared to vehicle controls ( Fig. 1A; p < 0.01), suggesting that endothelial cell senescence was reduced by senolytic treatment in vivo.
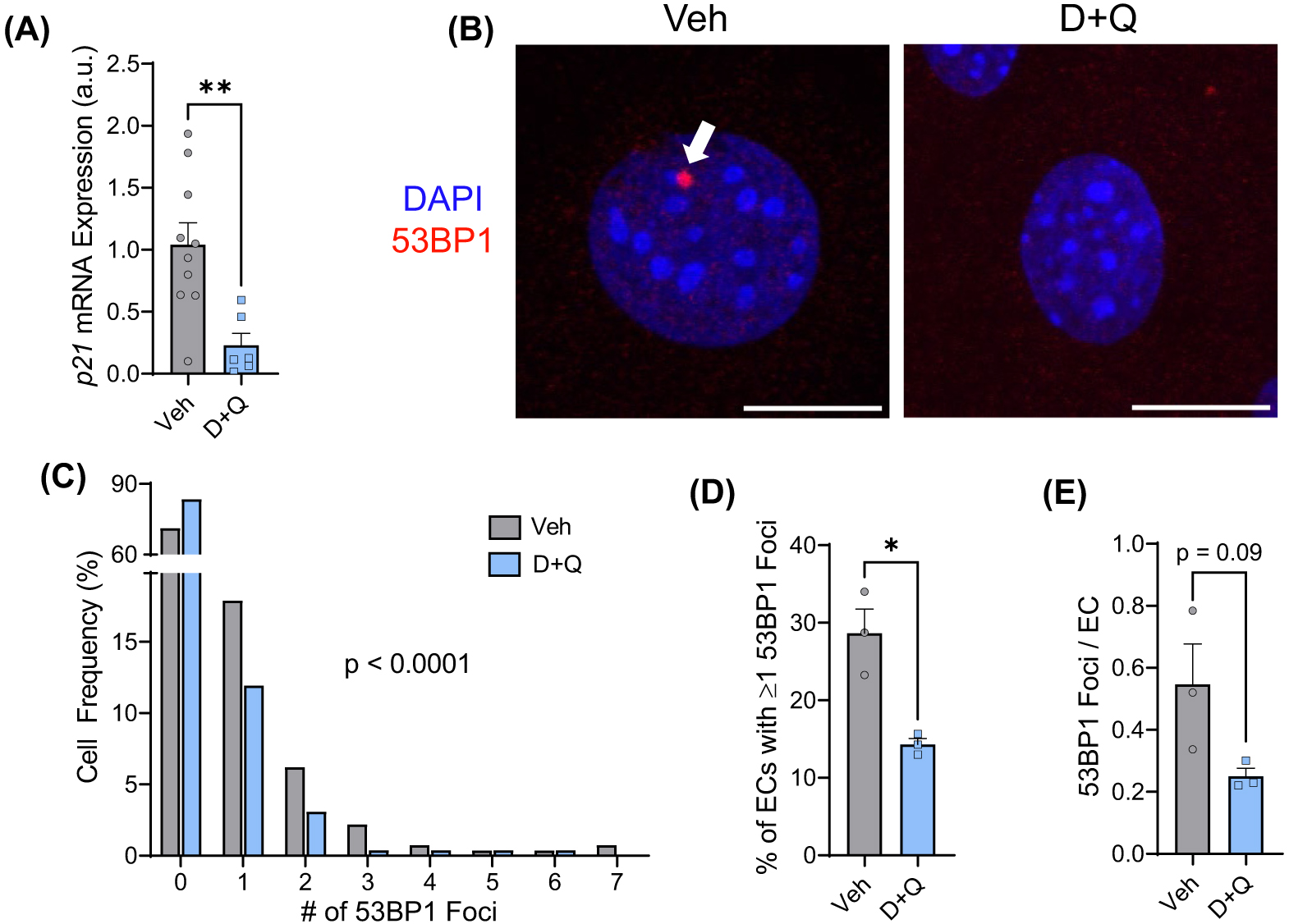
(A) Carotid artery endothelial cell enriched effluent mRNA expression of the cyclin-dependent kinase inhibitor and senescence marker p21. (B) Representative images of immunofluorescence for the DNA damage marker 53BP1, as indicated by the white arrow, in primary lung endothelial cells isolated from mice treated with a vehicle control (Veh) or the senolytic drug cocktail D+Q. (C) Frequency of 53BP1 foci in endothelial cells. (D) Percentage of endothelial cells with one or more 53BP1 foci. (E) The number of 53BP1 foci per endothelial cell. *p < 0.05, **p < 0.01, a.u.—arbitrary units, and scale bars—10 μm.
DNA damage is a robust inducer of senescence and is regarded as a hallmark of aging that can induce dysfunction independent of changes in cell fate choice. Therefore, we sought to determine if senolytic treatment affected the abundance of DNA damage in endothelial cells. The frequency of endothelial cells containing 53BP1 foci, a marker of DNA damage, was altered by senolytic treatment such that D+Q-treated mice had more endothelial cells without 53BP1 foci than vehicle-treated mice ( Fig. 1B,C; p < 0.0001). Furthermore, the percentage of endothelial cells containing 53BP1 was reduced ∼50% by D+Q (Fig. 1D; p < 0.05), and the number of DNA damage foci per cell tended to be reduced (Fig. 1E; p = 0.09). These data demonstrate that endothelial cells from mice treated with senolytics display reductions in a marker of senescence as well as the percentage of cells containing DNA damage.
Effect of senolytics on endothelial cell telomere dysfunction
Telomeres are repeated DNA sequences at the ends of chromosomes that are highly susceptible to damage. In addition, DNA damage at telomeres is persistent and thus highly predictive of senescence. Therefore, we sought to examine if D+Q reduced endothelial cell TIF (DNA damage signaling at telomeres). In comparison to vehicle-treated mice, D+Q-treated mice had more endothelial cells without TIF ( Fig. 2A,B; p < 0.01). Furthermore, D+Q mice displayed an ∼46% reduction in endothelial cells containing TIF ( Fig. 2C; p < 0.05) and an ∼58% decrease in the number of TIF per endothelial cell ( Fig. 2D; p < 0.05). Collectively, these data demonstrate that telomere dysfunction is less common in endothelial cells from mice treated with senolytics.
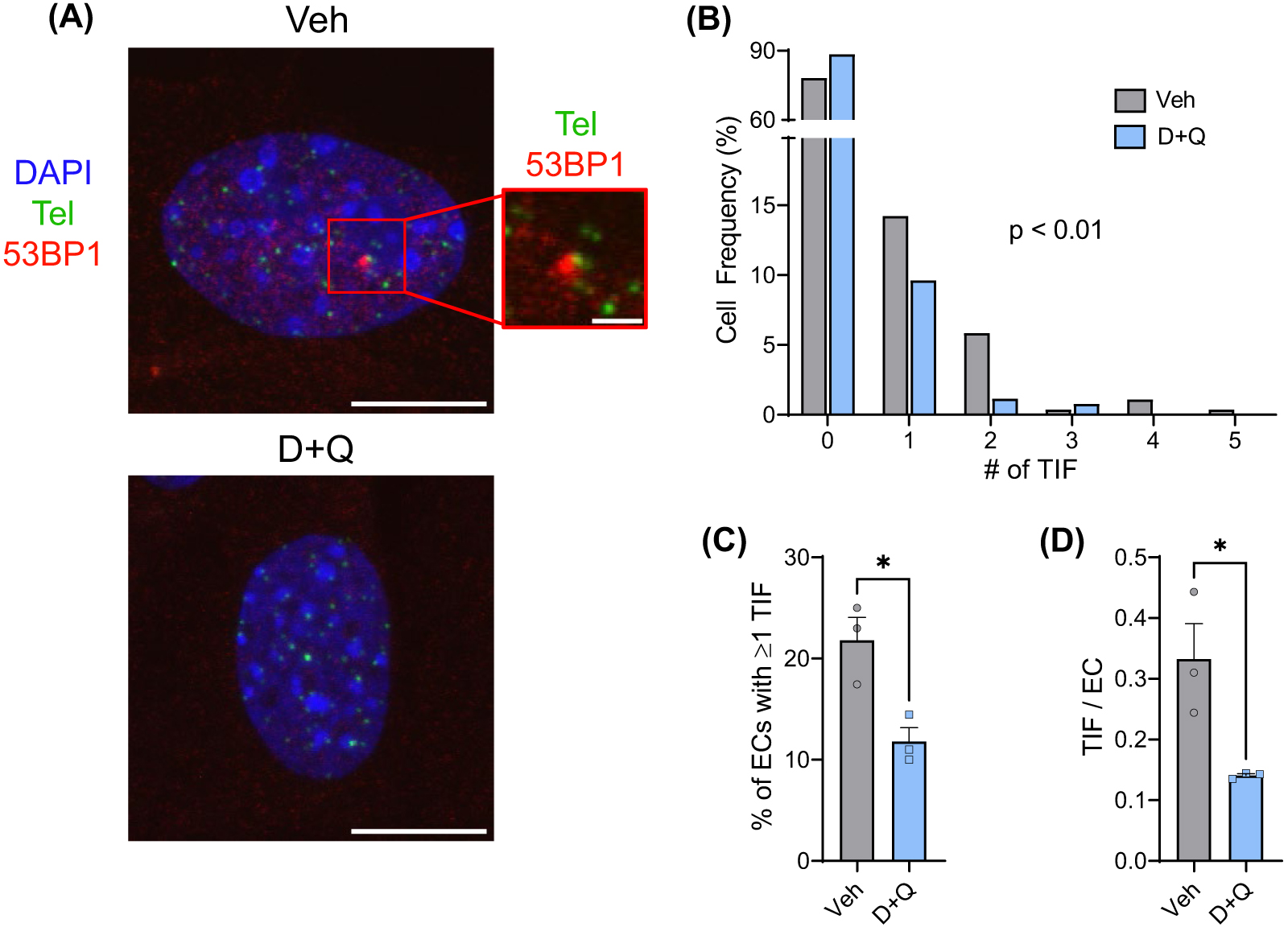
(A) Representative images of immunofluorescence fluorescent in situ hybridization (IF-FISH) for the DNA damage marker 53BP1 (red), and telomeres (Tel, green), in primary lung endothelial cells isolated from mice treated with a vehicle control (Veh) or the senolytic drug cocktail D+Q. Colocalization of 53BP1 and Tel signal is representative of a TIF. (B) Frequency of TIF in endothelial cells (ECs). (C) Percentage of endothelial cells with one or more TIF. (D) The number of TIF per endothelial cell. *p < 0.05 and scale bars—10 and 2 μm.
Effect of senolytics on endothelial cell telomere length
Telomeres shorten with each cell division and, when critically short, lose the ability to maintain the loop structure at their ends. This exposes telomere ends to the DNA damage response machinery, resulting in senescence. To assess whether senolytics impact telomere length, fluorescence intensity of endothelial cell telomeres was quantified. Interestingly, mean telomere length was reduced by ∼17% in endothelial cells of D+Q-treated mice ( Fig. 3A; p < 0.0001). Conversely, the frequency of mean telomere lengths was not different in endothelial cells from D+Q-treated mice ( Fig. 3B; p > 0.05) nor was there an appreciable alteration in the frequency of long or short telomere lengths ( Fig. 3C; p > 0.05). These data suggest that clearing senescent cells requires increased division of existing healthy cells that may result in a decrease in mean telomere length without influencing the frequency of critically short telomeres.
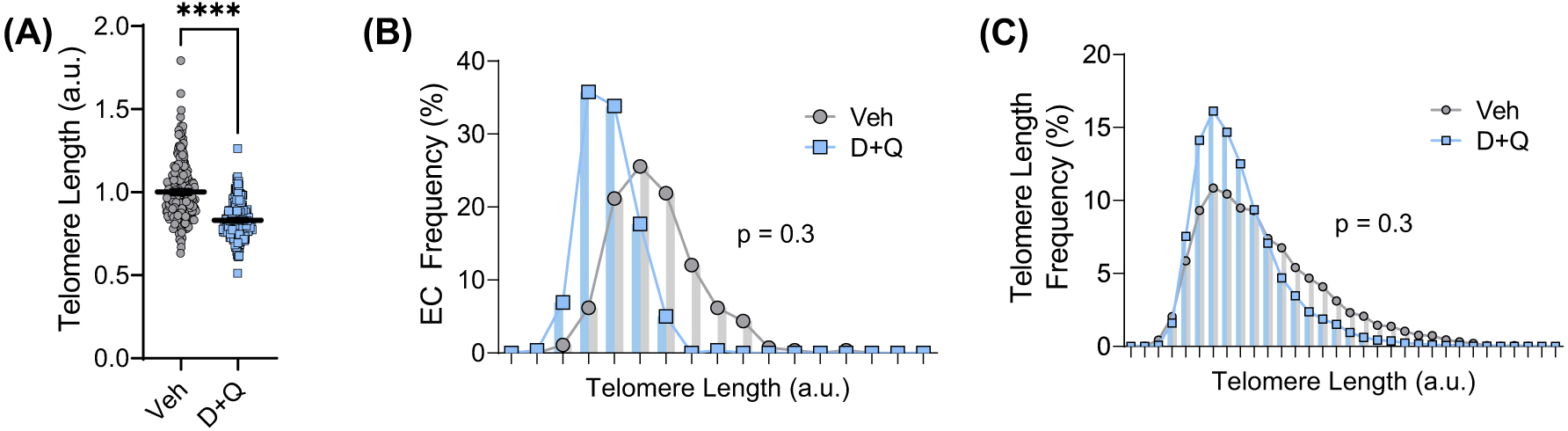
(A) Mean telomere length quantified from fluorescence intensity of IF-FISH in primary lung endothelial cells (ECs) isolated from mice treated with a vehicle control (Veh) or the senolytic drug cocktail D+Q. (B) Frequency distribution of endothelial cells based on mean telomere length. (C) Frequency distribution of telomere lengths. ****p < 0.0001 and a.u.—arbitrary units.
Next, we sought to elucidate the relationship between TIF and telomere length following treatment with D+Q. Mean telomere length was associated with the percentage of endothelial cells containing TIF ( Fig. 4A; R2 = 0.9, p < 0.01). Furthermore, mean telomere length was reduced in endothelial cells from D+Q-treated mice compared to vehicle-treated mice in cells with or without TIF ( Fig. 4B; p < 0.05); however, TIF status did not influence mean telomere length within each group ( Fig. 4B; p < 0.05). Maximum telomere length was reduced in endothelial cells from D+Q-treated mice with and without TIF, whereas minimum telomere length was greater in D+Q endothelial cells that did not contain TIF ( Fig. 4C; p < 0.05). The frequency of short telomeres was not different between cells with or without TIF in either vehicle-treated mice ( Fig. 4D; p > 0.05) or D+Q-treated mice ( Fig. 4E; p > 0.05). Taken together, these data support the notion that clearing senescent cells, which likely have TIF, increase the need for mitotic cell division resulting in decreased telomere length. However, reductions in mean telomere length are resultant of a reduction in maximal telomere length and thereby do not result in critically short telomeres.
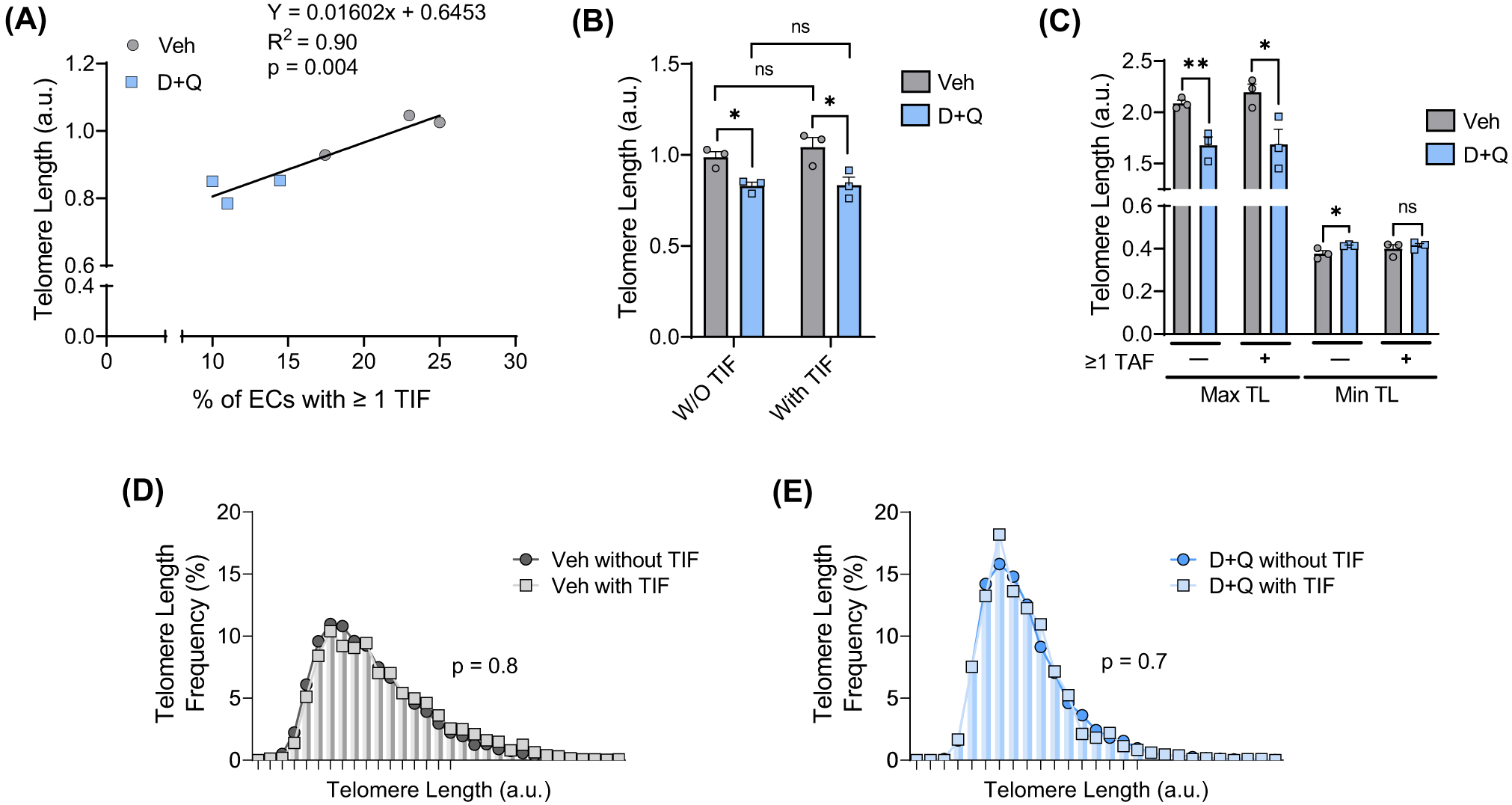
(A) Association between endothelial cell telomere length and the percentage of endothelial cells containing one or more TIF. (B) Mean telomere length in endothelial cells (ECs) with and without (W/O) TIF. (C) Maximum and minimum telomere length in endothelial cells with and without (W/O) TIF. (D) Frequency distribution of telomere lengths in cells with and without TIF from vehicle (Veh)-treated mice. (E) Frequency distribution of telomere lengths in cells with and without TIF from D+Q-treated mice. *p < 0.05, **p < 0.01, and a.u.—arbitrary units.
Discussion
The key novel findings of this study are as follows. First, treating aged mice with the senolytic cocktail D+Q reduces a marker of endothelial cell senescence. Second, senolysis reduces the percentage of endothelial cells containing DNA damage. Third, senolytics reduce the percentage of endothelial cells containing dysfunctional telomeres, as well as the amount of telomere dysfunction per endothelial cell. Finally, senolytics reduce telomere length, likely by increasing the need for cell replication through killing senescent cells. Reductions in telomere length induced by senolytics correspond with reductions in dysfunctional telomeres and are unlikely to induce further senescence because D+Q treatment does not result in critically short telomeres.
Several senolytic drugs were originally identified based on their ability to clear senescent endothelial cells4,5. Despite this, evidence that senolytics reduce endothelial cell senescence in old age is extremely limited14. In this study, we examined endothelial cell mRNA expression of the cyclin-dependent kinase inhibitor p21 that enforces senescence following DNA damage or telomere dysfunction6,8. As would be expected with a reduction in senescence, mice treated with D+Q displayed a reduction in endothelial cell p21 mRNA expression compared to their vehicle-treated counterparts. These findings are important because, prior to this study, a majority of the evidence demonstrating the efficacy of senolytics to clear senescent endothelial cells was derived from cell culture studies4,5. In addition, with advancing age, endothelial cells are one of the first cell types to become senescent7, and senescent endothelial cells have widespread physiological consequences including that they contribute to age-related arterial dysfunction and cardiovascular disease (CVD)6,8,15,16. Therefore, our study bolsters existing evidence that senolytics reduce endothelial cell senescence in advanced age.
Several molecular hallmarks of aging that are known inducers of senescence accumulate in endothelial cells with advancing age. For example, aging increases endothelial cell DNA damage and telomere dysfunction11,17, both of which can also induce dysfunction independent of changes in senescence12,18,19. In this study, examination of the impact of senolytics on endothelial cell DNA damage revealed that D+Q decreases the percentage of cells containing DNA damage. Not only is this important because DNA damage is a potent inducer of senescence17, but in addition, DNA damage contributes to age-related dysfunction through several diverse mechanisms. For example, DNA damage adversely impacts transcription and DNA replication, and can result in mutations that alter cellular function12. Consequently, mice with genetic reductions in DNA repair in endothelial cells exhibit vascular dysfunction as seen in advanced age20. In humans, genetic defects in DNA repair result in accelerated aging syndromes that often result in premature death due to CVD21,22. Similarly, genotoxic chemotherapy and radiation increase risk of CVD and death23–25. From a translational perspective, studies such as these highlight the importance of recognizing that senolytics reduce endothelial cell DNA damage.
Telomeres are the ends of chromosomes comprised of repeat sequences26. At the end of telomeres, a loop structure (t-loop) prevents chromosome ends from being recognized by the DNA damage response machinery26. Telomeres are sensitive to replicative and oxidative stress, both of which can inhibit t-loop formation, resulting in DNA damage response activation at telomeres (i.e., TIF)8. In this study, we found that senolytics reduce the percentage of endothelial cells containing TIF, as well as the number of TIF per endothelial cell. These findings are important because DNA damage at telomeres is persistent and TIF are highly predictive of senescence induction27,28. In mice, genetic induction of telomere dysfunction impairs endothelium-dependent vasodilation and results in an accelerated aging phenotype15,29. Furthermore, in humans, arterial TIF are associated with senescence and inflammatory signaling30. Telomere dysfunction can also have important effects beyond senescence including that it can influence gene expression of nearby genes18,31 and drive genomic instability19.
Recognition of telomere dysfunction as a hallmark of aging originates from the observation that telomeres shorten with each cell division, and when critically short, signal for senescence effectively limiting cellular lifespan in culture32. Evidence suggests that telomere dysfunction can also occur independent of changes in length11,30,32. To assess whether decreases in TIF were associated with changes in telomere length, we quantified telomere length. Interestingly, endothelial cell mean telomere length was reduced by ∼17% in D+Q-treated mice. Telomere length was strongly associated with the percentage of cells containing TIF. Based on the understanding that cells containing TIF are likely senescent, this indicates that clearing senescent endothelial cells requires cell division to replace cells killed by senolytics, leading to telomere attrition, or that D+Q directly shorten telomeres through an unknown mechanism. Importantly, we found that this reduction in telomere length was driven by shortening of the longest telomeres and did not result in critically short telomeres which could consequently increase the senescence burden. C57BL/6 mice have longer telomeres than humans33. Critically, short telomeres can contribute to increases in senescence burden, changes in gene expression in subtelomeric regions, and genomic instability18,19. Therefore, it would be of interest to determine whether the findings of this study translate to humans as clinical trials for senolytics continue.
In conclusion, this study provides important evidence on the effect of senolytics, including that they clear senescent endothelial cells in vivo. Moreover, we found that treatment with senolytics reduced hallmarks of aging including DNA damage and telomere dysfunction, both of which are robust inducers of senescence and can have additional deleterious consequences on cell and tissue function. These data indicate that clearing of senescent endothelial cells in old age leaves behind a population of cells that exhibit fewer features of vascular aging.
Acknowledgments
Funding: National Institutes of Health Awards R01 AG048366 (L.A.L.), R01 AG060395 (A.J.D.), 5K08AG070281 (E.T.), and 1F31AG076312 (S.I.B.). Veteran’s Affairs Merit Review Award I01 BX004492 (L.A.L.) from the US Department of Veterans Affairs Biomedical Laboratory Research and Development Service. The contents do not represent the views of the US Department of Veterans Affairs, the National Institutes of Health, or the U.S. Government.
Declaration of Interests
A.J.D. is a scientific adviser and stockholder, and L.A.L. is a stockholder in Recursion Pharmaceuticals. None of the work done with Recursion is outlined or discussed in this article.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.