Workshop Report
Biology of Stress Responses in Aging
Authors
Manolis Maragkakis,1,* Sulochan Malla,1 Maria Hatzoglou,2 Aleksandra Trifunovic,3 Adam B. Glick,4 Toren Finkel,5 Valter D. Longo,6 Susmita Kaushik,7 Pura Muñoz-Cánoves,8 Gordon J. Lithgow,9 Nirinjini Naidoo,10 Lauren N. Booth,11 Matthew J. Payea,1 Allison B. Herman,1 Rafael de Cabo,1 David M. Wilson 3rd,12 Luigi Ferrucci,1,* and Myriam Gorospe1,*
1National Institute on Aging Intramural Research Program, National Institutes of Health, Baltimore, MD, USA
2Department of Genetics and Genome Sciences, Case Western Reserve University, Cleveland, OH, USA
3Medical Faculty, Institute for Mitochondrial Diseases and Aging, University of Cologne, Cologne, Germany
4Department of Veterinary and Biomedical Sciences, Pennsylvania State University, University Park, PA, USA
5Aging Institute, University of Pittsburgh School of Medicine, Pittsburgh, PA, USA
6Longevity Institute and Davis School of Gerontology, University of Southern California, Los Angeles, CA, USA
7Department of Developmental and Molecular Biology, Albert Einstein College of Medicine, Bronx, NY, USA
8Centro Nacional de Investigaciones Cardiovasculares (CNIC), Madrid, Spain
9The Buck Institute for Research on Aging, Novato, CA, USA
10Chronobiology and Sleep Institute and Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
11Calico Life Sciences, San Francisco, CA, USA
12Biomedical Research Institute, Faculty of Medicine and Life Sciences, Hasselt University, Diepenbeek, Belgium
*Corresponding author: emmanouil.maragkakis@nih.gov, ferruccilu@grc.nia.nih.gov, myriam-gorospe@nih.gov
DOI:https://doi.org/10.59368/agingbio.20230001
Abstract
On April 28, 2022, a group of scientific leaders gathered virtually to discuss molecular and cellular mechanisms of responses to stress. Conditions of acute, high-intensity stress are well documented to induce a series of adaptive responses that aim to promote survival until the stress has dissipated and then guide recovery. However, high-intensity or persistent stress that goes beyond the cell’s compensatory capacity is countered with resilience strategies that are not completely understood. These adaptative strategies, which are an essential component of the study of aging biology, were the theme of the meeting. Specific topics discussed included mechanisms of proteostasis, such as the unfolded protein response (UPR) and the integrated stress response (ISR), as well as mitochondrial stress and lysosomal stress responses. Attention was also given to regulatory mechanisms and associated biological processes linked to age-related conditions, such as muscle loss and regeneration, cancer, senescence, sleep quality, and degenerative disease, with a general focus on the relevance of stress responses to frailty. We summarize the concepts and potential future directions that emerged from the discussion and highlight their relevance to the study of aging and age-related chronic diseases.
Introduction
Aging has been conceptualized as a continuous duel between damage accumulation that occurs from intrinsic or extrinsic stressors and resilience strategies that counteract this damage1. Stress responses have three general stages: (1) an alarm phase, (2) a resistance/recovery stage, and (3) exhaustion. The alarm phase involves the initial reactions of the body to stress, including cellular decisions related to “fight or flight,” whereas the resistance/recovery stage entails the activation of responses to cope and ultimately reverse the damaging effects of the stress. The last feature, exhaustion, occurs when the body’s ability to adapt to high stress levels eventually breaks down after either severe stress or a period of sustained stress that ultimately diminishes the adaptive response. Thus, an obvious question arises: are aging and chronic diseases the products of a gradual decline in the ability to cope with stress?
If stress is sufficiently severe, it will overwhelm the capacity of the cell to adapt and respond, leading to the activation of death mechanisms that contribute to degenerative pathologies2. However, stress responses might also become maladaptive with time, promoting systemic decline and the development of disease. Whether or not the failure of stress-response mechanisms at the cellular level is causally related to the phenotypic manifestation of aging is unknown and is a pressing topic for future research. Before highlighting the most recent advances outlined by the different speakers, we briefly summarize the key stress-response mechanisms that have been identified in mammals (Fig. 1). We close by identifying some of the main themes that emerged from the workshop and the directions of research deemed most critical looking to the future.
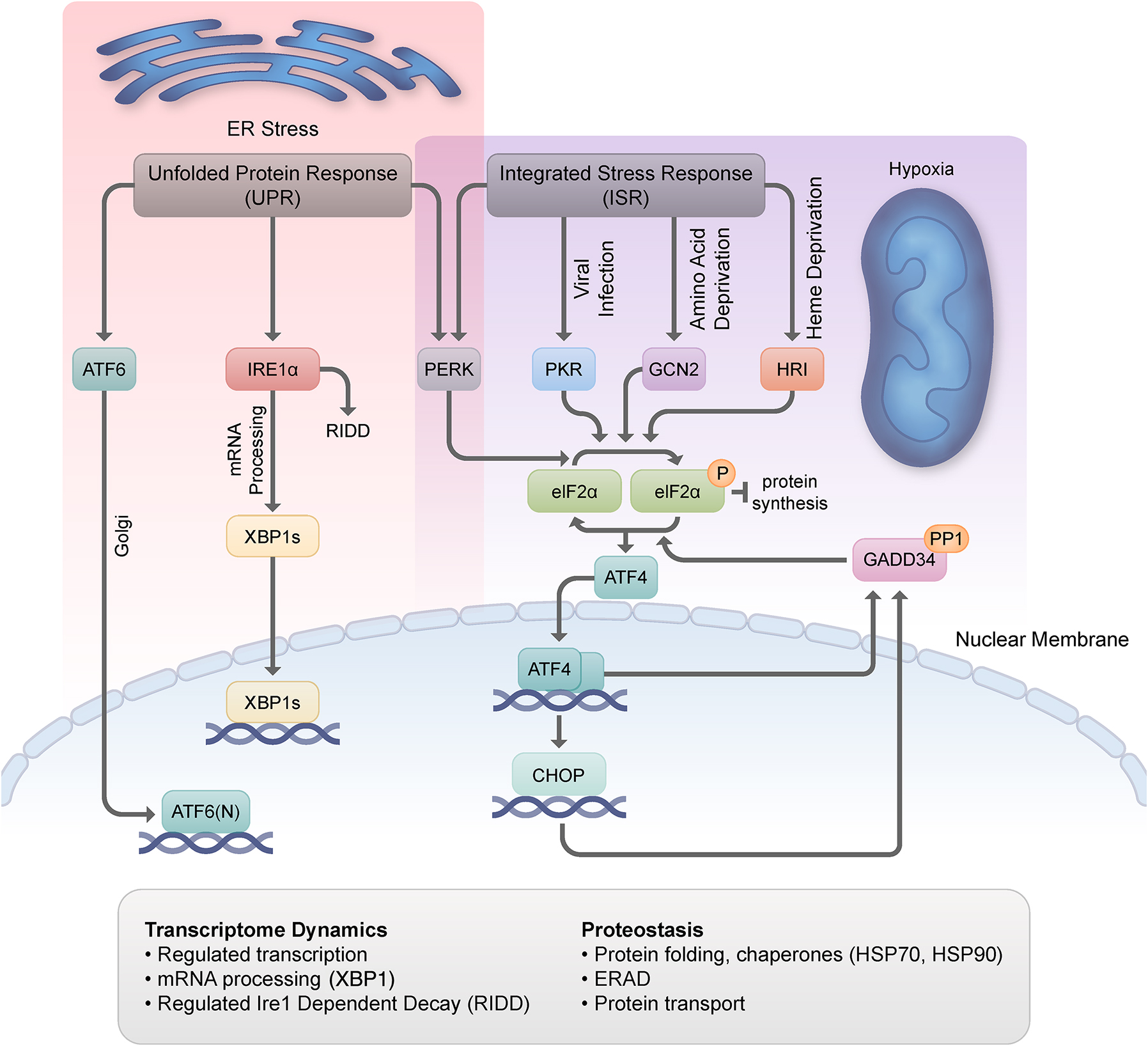
The accumulation of unfolded proteins in the endoplasmic reticulum (ER) can disrupt ER homeostasis and induces ER stress, with ensuing activation of the UPR to block the influx of more proteins until the balance is restored. The ER stress proteins are activating transcription factor 6 (ATF6), inositol-requiring enzyme 1 (IRE1) α, and protein kinase R (PKR)-like ER kinase (PERK). IRE1α precisely splices X-box-binding protein-1 (XBP1) mRNA, and the protein encoded by the cleaved mRNA is a transcription factor (XBP1s) that, together with ATF6, induces the transcription of UPR genes, including key chaperones. The PERK kinase phosphorylates the α subunit of eukaryotic translation initiation factor 2 (eIF2α) to inhibit protein synthesis and prevent further flux of proteins through the ER. eIF2α can also be phosphorylated by PKR in response to viral infection, general control non-derepressible 2 (GCN2) in response to amino acid starvation, and heme-regulated inhibitor (HRI) in response to heme deficiency. The general suppression of translation occurs concomitantly with the translation of select mRNAs, including that encoding the transcription factor ATF4. ATF4 in turn promotes the transcription of CCAAT/enhancer binding proteins (C/EBP) homologous protein (CHOP), also a transcription factor, and both transcriptionally induce the production of growth arrest- and DNA damage-inducible 34 (GADD34), a protein that in complex with the PP1 phosphatase, dephosphorylates eIF2α, restoring translation. Together, the UPR and the ISR jointly control proteostasis during the stress response; they modulate protein translation, protein folding, particularly through the action of chaperones such as HSP70 and HSP90, protein stability via ER-associated degradation, and protein transport. The UPR and the ISR also control transcriptome dynamics through processes like the selective transcription of mRNAs encoding stress-response proteins and via unconventional processing events such as specific cleavage of the XBP1 mRNA.
Unfolded protein response
The endoplasmic reticulum (ER) is a large, membrane-enclosed organelle that serves as a main site for protein synthesis and transport, protein folding, lipid and steroid synthesis, carbohydrate metabolism, and calcium storage. In situations that perturb ER homeostasis, such as Ca2+ depletion, hypoxia, altered glycosylation, or viral infection, unfolded proteins accumulate in the ER lumen and cause ER stress, triggering the unfolded protein response (UPR)3. The UPR aims to restore protein-folding homeostasis by (1) limiting protein synthesis and any additional strain on the folding system, (2) enhancing the removal of misfolded proteins via targeted degradation mechanisms, and (3) stimulating the production of chaperones and foldases that augment the overall folding capacity.
The UPR in mammalian cells is orchestrated by three principal ER transmembrane receptors: inositol requiring 1 (IRE1α), eukaryotic initiation factor 2α (eIF2α) kinase (protein kinase R [PKR]-like ER kinase [PERK]), and activating transcription factor 6 (ATF6). IRE1α is a type-I transmembrane protein that harbors a luminal sensor and cytoplasmic serine/threonine kinase and endoribonuclease domains4. Under conditions of ER stress, IRE1α appears to be activated by oligomerization and trans-autophosphorylation of IRE1 monomers. These modifications enhance its endoribonuclease function (its so-called regulated IRE1-dependent decay [RIDD] activity) and promote nonconventional cytoplasmic splicing of X-box-binding protein-1 (XBP1) mRNA, which encodes the bZIP transcription factor XBP1; XBP1 in turn transcriptionally induces the expression of proteins that promote protein folding and clearance of misfolded proteins from the ER5. ATF6α is an ER-localized, type-II transmembrane glycoprotein that dissociates from immunoglobulin-binding protein (BiP)/glucose-regulated protein 78 (GRP78) during ER stress, exposing two Golgi-localization sequences and triggering translocation of the protein to the Golgi apparatus6. There, ATF6α is cleaved by site-1 and site-2 proteases, releasing a 50-kDa amino-terminal fragment that enters the nucleus, binds to ER stress-response elements, and activates the expression of target genes that function to augment proteostasis. PERK, a type-I transmembrane protein and a member of the eIF2α kinase family7, reduces protein synthesis to relieve stress on the ER and participates in the integrated stress response (ISR, described below). When the UPR fails to reestablish proteostasis or remains activated for a long time, the response circuitry can trigger programmed cell death.
A subcomponent of the UPR, the so-called mitochondrial UPR (mtUPR), induces the production of chaperones and proteases that maintain mitochondrial protein homeostasis. The mtUPR is triggered by mitochondrial dysfunction, requires mitochondrial-to-nuclear communication, and engages as a central component the transcription factor, CCAAT-enhancer-binding protein homologous protein (CHOP). Most studies to characterize the mtUPR have been performed in worms and, therefore, our understanding of the mammalian mtUPR is in its early stages.
ISR
The ISR is a conserved cell survival pathway that responds to various internal and external inputs, insults, or disease states8. It can be activated by intrinsic stressors such as mitochondrial dysfunction and, similar to the UPR, ER stress caused by the accumulation of damaged, unfolded, or misfolded proteins. Main extrinsic stressors for the ISR include amino acid deprivation, glucose deprivation, heme deprivation, viral infection, hypoxia, oxidants, and oncogenic activation. The ISR engages four protein kinases that uniquely respond to stress signals and phosphorylate eIF2α, one of three subunits in the eIF2 complex: PERK, heme-regulated inhibitor (HRI), general control non-derepressible 2 (GCN2), and PKR. Phosphorylation of eIF2α suppresses the activity of the eIF2 complex, reducing global translation initiation and protein synthesis and allowing for the refolding of misfolded proteins that are crowding the ER. However, while eIF2α phosphorylation reduces general protein synthesis, it also promotes the translation of certain inhibitory upstream open reading frame (uORF)-containing transcripts that support survival, migration, and immune escape, most notably ATF4 mRNA. Whereas the ISR normally works to resolve stress by tamping down the energy-intensive cost of translation and preparing an adaptive response, in situations where the damage is excessive, the ISR can set in motion a cascade of events that lead to apoptosis. Moreover, defects in the ISR due to pathology or age can give rise to a maladaptive response that further promotes disease and drives the aging process.
Autophagy
Autophagy, which translates to “self-eating,” is an evolutionarily conserved preservation process that provides an organism with the ability to remove and recycle dysfunctional or damaged cellular components to avert disease development and restore biological function9. Autophagic responses are activated by many strains to the cell, including nutritional, oxidative, lipotoxic, proteotoxic, and genotoxic stresses. In mammalian cells, autophagy encompasses three major pathways: microautophagy, chaperone-mediated autophagy (CMA), and macroautophagy. Microautophagy involves direct endolysosomal action and the engulfment of cytoplasmic cargo via membrane invagination or protrusion10. Conversely, CMA is a highly selective degradation pathway, targeting soluble cytoplasmic proteins that are damaged (e.g., oxidized) or incorrectly folded, or functional proteins marked for selective degradation to terminate their activity11. Cytoplasmic CMA substrates are first recognized by a chaperone (heat shock protein HSP70), and the CMA-substrate/chaperone complex is then captured by LAMP2A, the lysosomal-membrane receptor that directs the binding, translocation, and degradation of the substrate protein within the lysosomal compartment. Macroautophagy involves the de novo formation of cytosolic double-membrane vesicles, that is, autophagosomes, to sequester and transport cargo, which ranges from proteins to aggregates to entire organelles, to the lysosome for bulk degradation12. Notably, evidence is emerging that there can be great selectivity in macroautophagy, with specific processes related to aggrephagy, lipophagy, ribophagy, mitophagy, pexophagy, reticulophagy, lysophagy, and xenophagy.
Stressors of different types, duration, and intensity can activate different mechanisms of autophagy. In the simplest terms, stressors that cause organelle damage are more prone to induce macroautophagy, whereas stressors that promote soluble protein damage elicit a CMA response. However, the different autophagic processes do not function in isolation13. For example, during periods of nutritional stress, macroautophagy is induced in the initial phases of starvation, whereas CMA activation follows at a later stage of prolonged starvation, because macroautophagy subsides. In situations of oxidative stress, macroautophagy is activated during acute, high-intensity challenges, whereas CMA is called into action during chronic, mild stress conditions. During times of lipotoxic stress, macroautophagy and CMA function in a coordinated manner, with the former handling degradation of small lipid droplets as a whole and the latter removing the lipid droplet coat proteins to make the stored lipids accessible to lipases and macroautophagy degradation (lipophagy). There is great need for further research to elucidate the rules that guide the coordinated and compensatory actions of autophagic responses.
It is well known that autophagy decreases with aging9,14. As a consequence, damaged proteins/organelles and macromolecular aggregates increase with age. The decreased macroautophagy function stems from the impaired induction of the autophagic machinery, defects in vesicular trafficking, and reduced fusion between the autophagosome and lysosome, culminating in the accumulation of autophagosomes and their associated cargo. In the case of CMA, reduced activity during aging stems from reduced LAMP2A levels on the lysosomes. For microautophagy, a defect in the pH of late endosomes is at least one factor in its age-related functional decline. Notably, artificially increased expression of either ATG5 or a mutant form of BECLIN1 (F121A) in mice from birth results in increased lifespan and improved healthspan and/or organ function (brain or heart)15,16. Similarly, enhanced CMA via increased expression of LAMP2A improves stress resistance and organ function in old organisms17. In humans, peripheral T cells from progeny of centenarians exhibit selectively higher macroautophagy activity that correlates with better immune function than their non-centenarian spouses18. Indeed, many geroprotectors and lifestyle interventions known to improve healthspan/lifespan upregulate autophagy, suggesting that enhancers of this cellular mechanism are promising therapeutic agents19. Accordingly, direct autophagic stimulators, such as Tat-Beclin1-peptide or compound C1 (Transcription Factor EB [TFEB]) for macroautophagy, or atypical retinoic acid receptor, alpha (RARα) inhibitors or humanin for CMA, were reported to be geroprotector compounds. Importantly, however, exaggerated macroautophagy can be detrimental to the cell, due to reduced mitochondrial mass and an overstressed mitochondrial pool leading to energy crisis and death, emphasizing the importance of finding the correct balance of autophagic upregulation to support healthy living and longevity. In targeting the LAMP2A receptor of CMA, there is no excess degradation of protein substrates, indicating that this pathway is an attractive therapeutic target.
Lysosomal stress response
In the 1950s, Christian de Duve identified “lysosomes” during his fractionation experiments that isolated membrane-bounded organelles containing acid hydrolases20. de Duve would later coin the terms “endocytosis,” “phagocytosis,” and “autophagy,” pathways that are now known to bring substrates for digestion into lysosomes. Beyond serving as intracellular “garbage disposals,” lysosomes play important roles in multiple pathways in eukaryotic cells, such as cell migration, gene regulation, metabolic signaling, cholesterol transport, and microbial killing. Accordingly, mutations in genes that encode enzymes localizing to the lysosome give rise to numerous pathologies. With age or stress, the lysosomal membrane is damaged and consequently permeabilized, permitting leakage of the lysosome content (namely hydrolytic enzymes and acidic conditions) into the cytosol. Although damaged lysosomes can be engulfed and cleared by healthy lysosomes (i.e., lysophagy), more recent findings reveal the existence of cytosolic protein complexes, known as the endosomal sorting complexes required for transport (ESCRT), which can be mobilized and recruited to lysosomes to repair damaged lysosomal membranes21,22.
Starvation, differential stress resistance, and differential stress sensitization
Early studies using yeast found that when cells are starved in low-glucose media, they display a phenotype of increased resistance to a range of stressors, such as the oxidizing agents hydrogen peroxide and menadione23. This enhanced resistance, which involves reallocating energy toward maintenance pathways and away from reproduction or growth processes, was demonstrated through genetic studies to be dependent on protooncogene orthologs, namely rat sarcoma virus (RAS), target of rapamycin 1 (TOR1), and cAMP-dependent protein kinase (PKA), which work coordinately to regulate the activity of stress-response transcription factors, such as MSN2/4 and GIS1. These observations set the stage for the potential of cancer cells to exhibit a differential stress resistance (DSR), as oncoprotein activity prevents the activation of protective mechanisms and stress resistance24. Consistent with this notion, short-term starved Saccharomyces cerevisiae or cells lacking protooncogene homologs were strikingly better protected against oxidative stress, chemical genotoxins, or chemotherapy drugs than cells expressing the oncogene homolog Ras2val19. Similarly, starvation or fasting of mice-bearing subcutaneous cancer resulted in increased survival following treatment with chemotherapeutic agents, such as cisplatin, etoposide, and doxorubicin, apparently through a mechanism that involves reduced glucose and insulin-like growth factor 1 (IGF1) signaling. Furthermore, dexamethasone and rapamycin, drugs that promote hyperglycemia and are widely used in cancer treatment, sensitize cardiomyocytes and mice to the cancer drug doxorubicin. Collectively, data from yeast, worms, flies, and mice, support the hypothesis that normal cells, but not cancer cells, respond to short-term starvation (reduced glucose and IGF1 signaling, downregulation of the RAS/AKT pathway, and activation of transcriptional responses) by entering a stress-resistant, resilience-like mode, giving rise to DSR and a therapeutic opportunity.
Regeneration and cellular senescence
Regenerative capacity refers to the ability of a cell, tissue, or organism to renew or restore the system back to its original state following a natural fluctuation or traumatic event that causes damage or a disturbance. It is well documented that the regenerative capacity of humans broadly decreases with age, due to various intrinsic and extrinsic factors25. The regenerative capacity of skeletal muscle, for instance, experiences a marked decline with age, as reflected by reduced generation of myofibers and increased fibrotic tissue upon muscle injury26. This loss in resilience has been tied to the impaired functionality of satellite cells, the resident skeletal muscle stem cells, and the onset of senescence as satellite cells differentiate during regeneration.
Cellular senescence, one of several cellular responses to stress, is the long-term arrest of damaged cells, induced by a variety of triggers, such as telomere shortening, genotoxic injury, mitogens, or inflammatory cytokines, which culminates in the activation of the p53 tumor suppressor and/or the cyclin-dependent kinase inhibitor p1627. Although senescence has shown beneficial roles in embryogenesis, wound healing, host immunity, and tumor suppression, mounting evidence indicates that occupation and accumulation of these non-proliferating cells in key cellular niches can have local and systemic pathological consequences. In addition to impairing tissue regeneration in muscle and other organs, senescent cells that accumulate with age secrete profibrotic proteins and proinflammatory factors (through the so-called senescence-associated secretory phenotype [SASP]), creating a toxic and damaging environment that further promotes age-related phenotypes.
Stress-Response Mechanisms at Work
Stress adaptation cycles and potential mechanisms for disease development
Maria Hatzoglou (Case Western Reserve University, OH, USA) introduced her talk in the context of dry eye syndrome, an example of a condition caused by increased osmolarity stress. In human corneal epithelial cells, moderate dehydration (400–500 mOsm) triggered an osmoadaptive response quite different from what was observed with severe dehydration (600 mOsm). Specifically, unlike the high-intensity response, osmoadaptation involved recovery of protein synthesis over time (i.e., encompassing alarm and resistance stages), suppression but not complete loss of mTOR activity, and increased production of growth arrest and DNA damage 34 (GADD34) and sodium-dependent neutral amino acid transporter-2 (SNAT2), with higher intracellular levels of amino acids. Osmoadaptation also included formation and eventual resolution of RNA-binding protein condensates through the activity of SNAT2, suggesting that amino acids act as chemical chaperones to facilitate reinitiation of translation and disassembly of protein aggregates. Collectively, exposure to subthreshold stress generated an osmoadaptation response that allowed reprogramming for survival, whereas severe hyperosmotic stress did not28–30.
An interesting question was then posed: how many cycles of stress can a cell experience and still retain the capacity to reestablish homeostasis and survival? In mouse pancreatic MIN6 β-cells, treatment for 24 hours with cyclopiazonic acid (CPA), a reversible sarcoplasmic/ER Ca2+-ATPase pump inhibitor, triggered a classic ISR with increased PERK activity, eIF2α phosphorylation, and higher ATF4 and GADD34 levels (Fig. 2A). In addition, protein synthesis decreased dramatically within the first hour (the alarm stage), yet recovered gradually over the next 24 hours (the resistance stage). After removing CPA, ISR markers returned to normal, and cells restored protein synthesis and preserved viability. When CPA was administered in repeated cycles, with brief recovery phases after removing the chemical stressor, transcriptome and proteome analyses revealed a chronic upregulation of pathways related to protein processing in the ER, proteasome, and protein export, and a chronic downregulation of pathways of insulin secretion and type-II diabetes mellitus, indicating compromised β-cell identity (Fig. 2A). Genes specifically affected in MIN6 β-cells were central to β-cell function (e.g., G6PC2 and ABCC8) and were found to be dysregulated in islet cells from type-I diabetes patients. Moreover, UPR signatures and β-cell identity markers distinguished healthy β-cells from β-cells of type-I diabetes patients. Hatzoglou concluded that β-cells experiencing periodic episodes of reversible ER stress can adapt and recover, preserving functionality, identity, plasticity, and homeostasis, whereas cells that suffer persistent, frequent periods of pathological ER stress ultimately lose adaptive plasticity and normal function31.
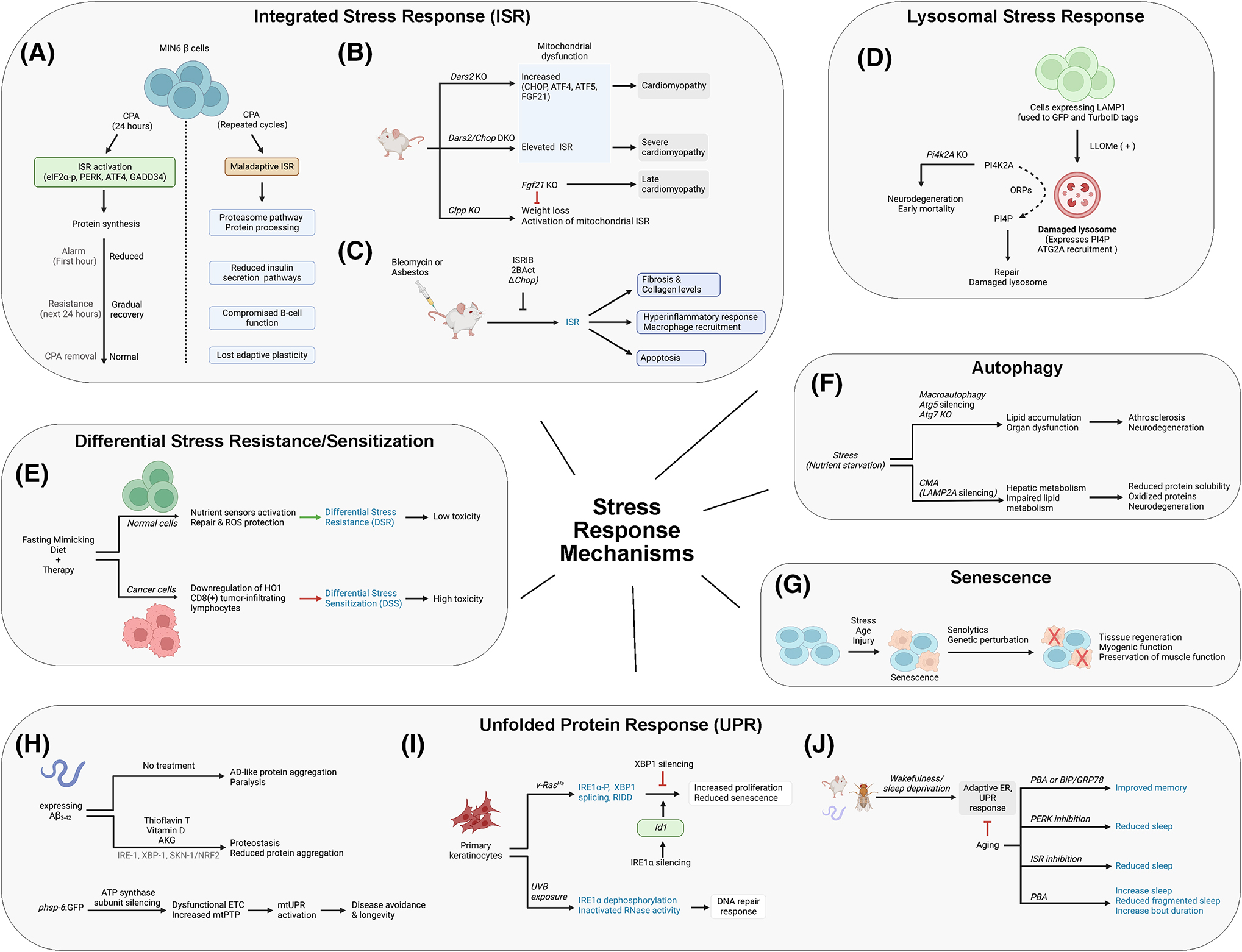
(A–C) Hatzoglou, Tifunovic, and Booth presented work on stress adaptation cycles and the potential mechanisms for disease development. They also discussed chemical modulation of the integrated stress response for potential therapies. (D) Finkel presented pathways for lysosomal stress and repair that are independent from the classic endosomal sorting complexes required for transport pathway. (E) Longo discussed differential stress sensitization (DSS) as a mediator for cancer therapy. (F) Kaushik presented selective autophagy in the context of the cellular and organismal response to stress and disease. (G) Muñoz-Cánoves discussed cellular senescence and the role of senolytics in tissue repair and aging. (H–J) Lithgow, Glick, and Naidoo discussed endoplasmic reticulum (ER) stress and the role of the unfolded protein response (UPR) in disease, neurodegeneration, and aging. They presented evidence for the chemical modulation of UPR as a potential avenue for disease intervention. For further information, see appropriate sections in text relevant to the different overview images.
Regulation of mitochondrial stress responses
Aleksandra Trifunovic (Institute for Mitochondrial Diseases & Aging, Cologne Graduate School of Ageing Research, Germany) focused on transcriptional reprogramming by mitochondrial toxins in mammalian cells, a molecular process that mimics key features of the ISR arm of the UPR. Tissue-specific ablation of mitochondrial aspartyl-tRNA synthetase (Dars2), encoding a key enzyme in the respiratory chain, allowed investigation of how impaired oxidative phosphorylation influenced key mediators of the ISR in mice. Ablation of Dars2 in the heart caused strong cardiomyopathy and early mortality by 8–9 weeks of age, with upregulation of CHOP, ATF4, and ATF5 in cardiomyocytes32, whereas Dars2/Chop double knockout mice died at 20 days of age, with severe cardiomyopathy and transcriptome alterations, particularly in genes related to the mitochondrial ISR (Fig. 2B). In addition, in a cell model lacking the mitochondrial complex IV assembly factor, cytochrome c oxidase assembly factor heme A:farnesyltransferase (COX10), ATF4 was elevated and ISR markers were observed, an effect that was suppressed by treatment with integrated stress response inhibitor (ISRIB), an inhibitor of the ISR. The data support the view that mitochondrial dysfunction results in ISR hyperactivation and that CHOP attenuates a prolonged response and consequent mitochondrial cardiomyopathy by modulating ATF4 production and downstream processes. Careful investigation of why ISR overactivation is detrimental revealed that mitochondrial dysfunction acutely reduces protein translation and subsequently reactivates cytoplasmic protein synthesis (despite an active ISR), culminating in an overwhelmed ER stress response and consequent disease development.
Fibroblast growth factor 21 (FGF21) is a stress-inducible cytokine mainly released from liver and adipocytes with critical roles in regulating energy balance and glucose and lipid homeostasis. FGF21 is also described as a “mitokine” because upon mitochondrial dysfunction, it is produced by tissues such as skeletal muscle, heart, and brain, where it appears to have endocrine and autocrine/paracrine activity. Genetic inactivation of Dars2 caused a robust increase in Fgf21 mRNA levels in the heart and FGF21 levels in circulation32. Employing a mild respiratory chain mutant model, that is, ClpXP protease (Cllp) knockout mice, whole-body FGF21 inactivation partially rescued the weight loss of single-mutant CLPP animals and decreased activation of the mitochondrial ISR specifically in heart (Fig. 1B). This combined effect, observed specifically in Clpp/Fgf21 knockout animals, engaged ATF5, Myc, and ER kinase (ERK) signaling, yet proceeded independent of classic ISR. In addition, the partial loss of the mitochondrial stress response in the Clpp/Fgf21 double knockout mice caused pathological cardiomyocyte remodeling defects with increased ventricle size and elevated heart-to-body ratios over time, culminating in the development of late cardiomyopathy. A broader implication of this work is that the degree of mitochondrial respiratory chain deficiency can influence the type of ISR and the associated molecular pathways33.
Cellular and organismal impact of the ISR
Lauren Booth (Calico Life Sciences, CA, USA) discussed ongoing efforts to understand the role of a maladaptive ISR in age-related disease and loss of tissue homeostasis as well as the potential therapeutic benefits of ISR attenuation. Using computational tools and publicly available gene expression datasets, scientists at Calico have identified disease states such as pulmonary fibrosis and amyotrophic lateral sclerosis (ALS) as being associated with an aberrantly high-ISR signature. An ongoing clinical trial (NCT04948645) with ALS-affected individuals aims to determine whether attenuation of the ISR serves as an effective therapeutic strategy.
Recent preclinical efforts have focused on idiopathic pulmonary fibrosis (IPF), a chronic and progressive disease that is more prevalent in the older population. IPF is thought to be triggered by damage to the alveolar epithelia from stressors that are known to induce the ISR and IPF, and it is characterized by hallmarks of a maladaptive ISR: increased cell death, inflammation, and macrophage recruitment34. Accordingly, suppression of the ISR via administration of ISRIB or 2BActivators (2BAct) reduced fibrosis and collagen levels in young and old preclinical mouse models that employed bleomycin or asbestos to induce injury (Fig. 2C)35. Moreover, attenuation of the ISR via ISRIB or 2BAct suppressed the hyperinflammatory response and macrophage recruitment in the lung disease models35. Inactivation of the ISR via genetic deletion of the pro-apoptotic, ISR-activated transcription factor CHOP or suppression of the ISR by ISRIB also reduced the accumulation of apoptotic cells at the site of injury (Fig. 2C)35,36. In pulmonary fibrosis, epithelial repair by alveolar type II (AT2, stem cells) to alveolar type I (AT1, gas exchange cells) differentiation stalls in a Krt8+ transition state37,38 that is characterized by a high-ISR gene expression signature (Kimmel J., 2021, unpublished). Consistent with a role for a maladaptive ISR in the loss of alveolar epithelial tissue homeostasis, this AT2 to AT1 differentiation defect can be corrected by ISRIB35,39, potentially supporting recovery and repair in the affected region. The data collectively support the ideas that (1) a chronic or maladaptive ISR can disrupt tissue homeostasis by increasing cell death and inflammation and by altering differentiation trajectories and (2) attenuation of this maladaptive ISR can serve as a target for therapeutic intervention.
Lysosomal stress and repair
The talk by Toren Finkel (University of Pittsburgh, PA, USA) began in the context of ESCRT, a complex that is mobilized and recruited to repair damaged lysosome membranes21,22. Given that rapid lysosomal membrane repair is only modestly impeded in the absence of critical ESCRT components, his team searched for ESCRT-independent repair systems. Using a proximity biotinylation approach, where lysosomal-associated membrane protein 1 (LAMP1) was fused to green fluorescent protein (GFP) and TurboID tags and stably expressed in human cells, they identified proteins recruited to damaged lysosomes following exposure to the lysosome toxin L-leucyl-L-leucine methyl ester (LLOMe). Proteins enriched on the surface of damaged lysosomes included ESCRT components and many factors involved in lipid signaling pathways. Specifically, the lipid phosphatidylinositol 4-phosphate (PI4P) was observed on the surface of the lysosome, whereas in untreated cells, PI4P was almost exclusively found in the Golgi complex. Gene-editing experiments revealed that generation of PI4P on the lysosome surface and rapid lysosomal repair were dependent on the kinase activity of phosphatidylinositol 4-kinase type 2 alpha (PI4K2A) (Fig. 2D). Based on their proteomic screen, Finkel and colleagues hypothesized that the de novo synthesis of PI4P by PI4K2A would be linked to the recruitment of the oxysterol-binding protein (OSBP)-related proteins (ORPs) to the surface of damaged lysosomes. ORPs are a family of eukaryotic sterol and phosphoinositide-binding and transfer proteins that contain a lipid-binding domain and facilitate the tethering of organelles to the ER and exchange of PI4P for other lipids, such as cholesterol or phosphatidylserine. Consistent with the proposed model, ablation of either PI4K2A or ORP9, 10, 11, and OSBP reduced lysosome-ER contact following damage by LLOMe (Fig. 2D).
Additional studies revealed that the predominant lipid exchanged for PI4P following ER-lysosomal tethering is phosphatidylserine. ATG2A, a lipid transporter shown to be activated by phosphatidylserine and to mediate autophagosomal membrane expansion, is also recruited to lysosomes upon damage, where the enzyme supports phosphatidylserine-stimulated bulk transfer of lipids for subsequent membrane stability and repair. There appears to be an ancillary backup pathway that engages OSBP and the transfer of cholesterol to further support membrane reconstitution. Interestingly, Pi4k2a knockout in mice causes early mortality that arises mainly from neurodegeneration associated with massive lysofuscin accumulation, potentially supporting a broader role for lysosomal defects in age-related degenerative diseases40 (Fig. 2D). Moreover, inactivation of the PI4K2A pathway results in increased Tau spreading in cell models, reaffirming the importance of the identified mechanism in preserving normal lysosome operations. In aggregate, these data indicate that phosphoinositide critically contributes to preserving the lysosomal membrane, with important consequences on age-related diseases characterized by impaired lysosomal function.
DSS in normal and cancer cells
Following earlier themes in the workshop, Valter Longo (University of Southern California, CA, USA) focused his talk on the apparent differential stress sensitization (DSS) of cancer cells24. Pre-clinical and clinical strategies have been developed around a periodic fasting mimicking diet (FMD) that (1) entails low calories, low sugar, low protein, and high unsaturated fats; (2) targets the essential signaling pathways underlying the DSR and DSS; and (3) is more agreeable to clinicians and potentially patients. In mice, the combination of fasting/FMD and chemotherapy led to a robust increase in cancer-free survival in models of lung and triple-negative breast cancer, in keeping with a role of calorie restriction as a differential enhancer of stress-response mechanisms. In addition, the combination of chemotherapy and FMD enhanced T-cell-dependent targeted killing of cancer cells by promoting the actions of cytotoxic CD8(+) tumor-infiltrating lymphocytes (Fig. 2E). This FMD-mediated cancer cell sensitization was partially enabled by downregulation of the stress-responsive enzyme heme oxygenase-1 (HO1)41. Similarly, adding FMD to vitamin C treatment reverses the increase in HO1 caused by vitamin C alone, resulting in increased reactive iron, reactive oxygen species, and cell death in KRAS-mutated colorectal cancer mouse models; these effects were further potentiated by chemotherapy42. Because data mining of the Cancer Genome Atlas indicates that colorectal cancer patients with mutated KRAS display longer overall survival, the current results suggest that the combination of FMD and vitamin C represents a promising low-toxicity intervention against colorectal cancer and other KRAS-mutated tumors, which could be particularly important for the frail elderly at risk for high-grade adverse events caused by a variety of standard therapies.
In a separate study, periodic fasting or FMD enhanced the efficacy of endocrine therapeutics tamoxifen and fulvestrant in the treatment of hormone receptor-positive breast cancer through a mechanism that involved lower circulating IGF1, insulin, and leptin, as well as inhibition of AKT–mTOR signaling via upregulation of EGR1 and phosphatase and tensin homolog (PTEN)43. In contrast, starvation of triple-negative breast cancer cells led to the activation of escape pathways, notably CycD–CDK4/6 and PI3K-AKT, and downregulation of CycB–CDK1. Accordingly, the combination of FMD and inhibitors targeting mTOR, PI3K, and AKT, coupled with FMD prior to treatment, reversed cancer growth and sustained regression, with the latter occurring primarily by reducing the numbers and stemness of cancer stem cells44. In ongoing separate clinical trials, fasting/FMD cycles appear to ameliorate the chemotherapy-induced decrease in the quality of life, improve the clinical and corresponding pathological response of HER2-negative stage II/III breast cancer patients, and have no effect (or sometimes a beneficial effect) on muscle mass/strength45. These positive outcomes underscore the potential for DSS in the treatment of a variety of cancers, based on the consistent observation that cancer cells, by disobeying starvation response orders, not only fail to become protected but also often enter an unsustainable metabolic mode, characterized by mutations and the lack of nutrients and factors required to sustain the metabolic rewiring (e.g., the increased glucose requirement associated with the Warburg effect). Thus, mouse and cellular work support the potential for the combination of periodic FMD cycles with a wide variety of cancer treatments, ranging from chemotherapy to radiotherapy, kinase inhibitors, hormone therapy, and immunotherapy, to generate high toxicity to cancer but not normal cells (Fig. 2E).
Selective autophagy in the cellular and organismal response to stress
Susmita Kaushik (Albert Einstein College of Medicine, NY, USA) centered on the contribution of different autophagic pathways to stress response. Inactivation of macroautophagy by silencing Atg5 in cells or by ablating the Atg7 gene in mouse liver resulted in lipid accumulation and organ dysfunction following nutrient deprivation, suggesting that macroautophagy is essential for removing excess lipid droplets46 (Fig. 2F). Inactivation of CMA via conditional Lamp2a gene deletion in liver caused hepatic glycogen depletion and hepatosteatosis, indicating a role for this process in regulating hepatic metabolism as well, likely via the targeted degradation of key enzymes involved in carbohydrate and lipid metabolism47. Similarly, engineered defects in either macroautophagy or CMA in mice resulted in atherosclerosis, presumably through distinct mechanisms that culminate in impaired lipid metabolism48,49. In the context of proteotoxicity, inactivation of macroautophagy via conditional Atg7 knockout in neurons triggered neurodegeneration and the accumulation of ubiquitin-positive aggregates in the affected brain regions50,51. A similar neurodegenerative phenotype, albeit with different kinetics, was also observed in mice lacking LAMP2A, which displayed pathological features of reduced protein solubility and high levels of oxidized proteins and ubiquitin-positive aggregates52 (Fig. 2F). Crossing the CMA-null mouse (lacking LAMP2A) with an Alzheimer’s disease (AD) mouse model (3xTg) further supported the conclusion that CMA plays an important role in mitigating neurodegeneration, because CMA-null 3xTg mice exhibit more rapid protein aggregation pathology (e.g., Tau) in comparison to the 3xTg mice alone52.
Interestingly, inactivation of macroautophagy via deletion of the Atg5 gene led to a general upregulation of CMA in mouse embryonic fibroblasts53, while, conversely, silencing LAMP2A in NIH3T3 fibroblasts increased macroautophagy54, suggesting a reciprocal compensation between the two autophagy pathways. However, their relationship is complex, and compensation seems to be cell type and context dependent. For instance, CMA inactivation is not accompanied by an increased macroautophagy response in brain52 or hematopoietic stem cells55. In retinal cells, while suppression of macroautophagy leads to enhanced CMA, the reverse is not observed in situations of CMA inactivation56. Resting T cells in which CMA has been eliminated exhibit upregulation of macroautophagy, but activated T cells do not57. Moreover, while liver typically exhibits normal compensatory responses, in situations involving an aged liver or a chronically stressed liver (e.g., from high-fat diet), CMA inactivation is not compensated for by increased macroautophagy activity58. Interestingly, whereas compensation between macroautophagy and CMA occurs in response to blockage of one of the two pathways, at present, there is no evidence that upregulating any of the autophagic pathways leads to reduced function of the other(s).
Cellular senescence in tissue repair and aging
Pura Muñoz-Cánoves (Universitat Pompeu Fabra, Spain) explained that the ability to track and study cellular senescence in vivo is complicated, mostly due to the rarity of the event and the difficulty in unequivocally detecting senescent cells. To facilitate this task, her team isolates senescent cells from their biological native environment using p16-3MR mice59. These animals harbor the senescence-responsive promoter of p16Ink4a driving expression of the 3MR, (trimodality reporter) fusion protein; 3MR contains functional domains of a synthetic Renilla luciferase (LUC), monomeric red fluorescent protein (mRFP), and a truncated herpes simplex virus 1 (HSV-1) thymidine kinase (HSV-TK) that converts ganciclovir into an apoptosis inducer. After inducing muscle injury, senescent cells were observed to accumulate at low levels in both young and old mice. In old mice, this accumulation was more pronounced over time post-injury, likely due to poor clearance of senescent cells. The reduction of senescent cells via genetic mechanisms improved regeneration in both young and old mice, with muscle strength improving concomitantly. Application of the senolytic mixture Dasatinib + Quercetin (D + Q)60, had a similar beneficial effect on myogenic regeneration and preservation of muscle function (Fig. 2G).
Following isolation of senescent cells, which comprised ∼2%–3% of the total population using Fluorescence-activated Cell Sorting (FACS), single-cell RNA-sequencing studies were performed to compare changes in gene expression patterns. Some notable features included: high-transcriptomic heterogeneity among senescent cells, even within the same muscle niche; a general downregulation of transcription and translation factors, particularly those involved in pathways related to fibrosis and inflammation; increased expression of SASP factors; and secretion of inflammatory and matrix-remodeling proteins. In addition, despite the broad changes in gene expression programs, many of the lineage traits (for satellite and fibro/adipogenic progenitors) were retained. In sum, the talk by Muñoz-Cánoves underscored the impact of senescent cells on the ability of skeletal muscle to restore homeostasis following damage.
Chemical modulation of stress response during aging
Gordon Lithgow (The Buck Institute for Age Research, CA, USA) discussed the influence of life-extending compounds (specifically thioflavin T, α-ketoglutarate [AKG], and vitamin D) in the free-living transparent nematode Caenorhabditis elegans. These compounds appear to have similar beneficial effects in age-related parameters in rodents; for example, AKG compresses morbidity in mice, suppresses graying of the hair, and increases healthspan relative to control animals61. Notably, all three compounds appear to enhance proteostasis mechanisms in C. elegans. For example, worms expressing peptide Aβ3-42 exhibit AD-like protein aggregate pathology, eventually becoming paralyzed, and this phenotype is prevented by thioflavin T62. A similar protective effect, to a lesser magnitude, was observed for AKG and vitamin D, indicating shared mechanisms of action that involve reducing the aggregation of toxic proteins (Fig. 2H, top). Interestingly, a broader look at chemicals that promote lifespan extension indicate that all are able to suppress Aβ-driven paralysis in worms. Protein mass spectrometry analysis of captured insoluble material that accumulates with age in worms revealed the presence of proteins related to the tricarboxylic acid cycle (TCA) cycle, oxidative phosphorylation, the translational machinery, RNA-binding proteins, and stress granules63. In addition, artificial expression of the toxic Aβ peptide induced a similar pattern of protein aggregation, albeit in an accelerated manner, consistent with the notion that aging pathologies share underlying molecular processes. Conversely, treating worms with vitamin D dramatically reduced the age-dependent accumulation of insoluble proteins through a mechanism requiring IRE-1, XBP-1, and SKN-1/NRF2, despite no evidence that the UPR was induced (Fig. 2H, top).
The mitochondrial permeability transition pore (mtPTP) is a nonspecific transmembrane channel that resides in the mitochondrial inner membrane. Stresses that cause a rise in Ca2+, adenosine depletion, or oxidative damage trigger an opening of the mtPTP leading to enhanced permeability of the mitochondrial membrane, swelling of the organelle, and cell death. There is evidence that mtUPR activation is associated with both disease avoidance and longevity and with disease or stress propagation. Using an intestinal reporter worm that harbors a phsp-6:GFP cassette to track mtUPR activity, the effect of disrupting the electron transport chain (ETC) on mtUPR activation was examined during development and in adulthood64. Consistent with earlier studies, defective ETC induced by silencing a subunit of the F-type ATP synthase (complex V) during development resulted in activation of the mtUPR. Conversely, impairment in ETC function during adulthood by silencing other mitochondrial proteins did not activate the mtUPR, except when complex V activity was disrupted by atp-3 silencing. Moreover, unlike the life extension seen upon ETC inactivation and consequent induction of the mtUPR via atp-3 targeting during development, atp-3 silencing in adulthood shortened lifespan, despite activation of the mtUPR. Further analysis revealed that mtPTP activity is necessary for mtUPR induction and the reduction in lifespan observed in adult worms, revealing a complex mtPTP/mtUPR nexus at the core of disease processes and aging (Fig. 2H, bottom).
Importance of the ER stress sensor IRE1α in response to oncogenic and environmental stress
Adam Glick (Pennsylvania State University, PA, USA) discussed associations between UPR activation and survival and adaptation of cancer cells. To determine the role of IRE1α and ER stress responses in the early stages of cancer, studies were performed by exposing mouse skin to the carcinogen 7,12-dimethylbenz[a]anthracene (DMBA), complemented by studies in a cell culture model of epidermal keratinocytes infected with a v-RasHa retrovirus, which induces transient hyperproliferation, followed by senescence. Grafting of the hyperproliferative cells onto mouse skin leads to a benign papilloma, the first step in malignant progression. Introduction of oncogenic v-RasHa into primary mouse keratinocytes induced expansion of the ER and upregulation of UPR target genes65. Given that the observed ER stress was transient and diminished as cells entered the senescent phase in culture, the impact of proteostatic stress was determined by application of 4-phenyl butyrate (PBA), a known ER stress suppressor. Treatment of v-RasHa-transduced primary keratinocytes with PBA reduced proliferation and increased cellular senescence, implicating ER stress in growth promotion. In addition to ER stress, v-RasHa-induced expression and phosphorylation of IRE1α without activating PERK or ATF6 transiently increased XBP1 splicing and elevated RIDD activity. Whereas XBP1 silencing suppressed proliferation and promoted senescence in v-RasHa-transduced primary keratinocytes, knockdown of IRE1α increased proliferation and prevented senescence (Fig. 2I). Keratinocytes isolated from Xbp1−/− mice exhibited a similar pattern of increased senescence, whereas silencing IRE1α in Xbp1−/− cells reduced senescence. Collectively, these data indicate that RIDD activity is necessary for Ras-induced senescence, and XBP1 is important for the proliferative response. Id1 mRNA, encoding ID1 (inhibitor of DNA binding 1, a helix-loop-helix [HLH] transcription factor), was among those mRNAs that were downregulated by v-RasHa transduction but restored upon IRE1α silencing but not XPB1 silencing. ID1, which lacks DNA-binding activity and inhibits the DNA-binding and transcriptional activation of basic HLH proteins with which it associates, is implicated in cell proliferation, senescence, and differentiation. Expression of v-RasHa in primary keratinocytes reduced Id1 mRNA and ID1 levels, and ID1 silencing in Ras-activated IRE1α-deficient keratinocytes restored senescence (Fig. 2I).
Ultraviolet (UV) light is the major cause of skin cancer in humans and, therefore, delineating the effect of UV exposure on stress responses is of great interest. UV irradiation generates pre-mutagenic DNA damage forms including cyclobutane-pyrimidine dimers (CPDs), as well as reactive oxygen species and ER stress. Unexpectedly, UVB irradiation of mouse keratinocytes transiently dephosphorylated IRE1α and inactivated its RNase activity. Moreover, IRE1α-deficient keratinocytes were hypersensitive to UVB exposure both in a mouse model that exhibited UV-related sloughing of the epidermis, and in two culture models. In addition, IRE1α-deficient cells displayed increased apoptosis and CPD accumulation following UV irradiation, implying an impaired DNA repair response (Fig. 2I). Consistent with this conclusion, removal of UV-induced CPDs is slower in IRE1α knockdown cells, which also exhibit lower ATR phosphorylation and generation of γH2AX foci, markers of replicative stress and double-strand breaks, respectively. In sum, this work identifies a role for IRE1α in promoting the DNA damage response following exposure to UV irradiation.
The role of ER stress in age-related sleep/wake fragmentation
The talk by Nirmala Nirinjini Naidoo (University of Pennsylvania, PA, USA) began with the message that adequate quantity and quality of sleep is important for cognition, memory, mood, mental health, and physical aptitude, as well as cardiovascular, cerebrovascular, and metabolic health. Conversely, insufficient or poor-quality sleep is associated with an increased risk of mortality and contributes to individual risk of dementia, cardiovascular disease, diabetes, obesity, and cancer66. In particular, poor sleep has been associated with amyloid deposition, although it is not currently known if poor sleep causes the misfolding and insolubilization that triggers the UPR and the ISR67 (Fig. 2J). Understanding the molecular pathways that underlie healthy sleep can help identify interventions to minimize the harmful consequences of sleep deprivation, circadian misalignment, and sleep disorders.
In mice exposed to different periods of wakefulness, key markers of ER stress and the UPR were elevated with longer sleep deprivation in the cerebral cortex; UPR was also elevated after acute sleep loss in flies, birds, and rats, pointing to a conserved, universal stress response. In aged mice, this adaptive response is diminished or lost, with a coincident increase in GADD34 and proapoptotic proteins such as activated caspase-12; the same aged mice exhibit chronic activation of the UPR (i.e., a maladaptive UPR), with higher levels of CHOP and GADD34 in the nucleus, particularly in brain regions relevant to wake maintenance and alertness and susceptible to neurodegeneration (i.e., the locus coeruleus and orexin neurons, respectively)68 (Fig. 2J). Conversely, BiP/GRP78 expression in both fly and mouse brain decreases with age, whereas treatment with PBA, a chemical chaperone known to suppress ER stress and extend lifespan, increased the total amount of sleep of aged flies and reduced fragmented sleep69. Aged mice administered PBA similarly exhibited reduced number of sleep and wake bouts and increased bout duration, consistent with sleep/wake consolidation. In aged mice, PBA reduced PERK phosphorylation and ATF4 activity in cortical and hippocampal tissue samples, suggesting a suppression of the UPR, along with lower GADD34 activation, and a corresponding increase in AKT and CREB phosphorylation, the latter of which plays a role in BDNF induction and memory consolidation. PBA treatment or overexpression of BiP/GRP78 improved the performance of old mice in memory tests70 (Fig. 2J).
Pharmacological inhibition of PERK via GSK2606414 reduced sleep in both Drosophila and zebrafish, indicating an evolutionarily conserved role for the kinase pathway in facilitating sleep71. Genetic knockdown of PERK also reduced sleep in Drosophila. Similarly, activation of eIF2β with ISRIB and consequent stimulation of protein translation reduced sleep in flies, as did administering tunicamycin (an inhibitor of protein glycosylation that induces ER stress) to young flies71 (Fig. 2J). Collectively, these data indicate that sleep defects caused by aging or by pharmacological or genetic inducers, are associated with ER stress and the UPR, and that such stress-response mechanisms could potentially serve as powerful therapeutic targets.
Closing Considerations
The evolutionary selection of resilience mechanisms that work to resolve macromolecular damage and enable survival was fundamental to the extension of longevity. The core resilience platform involves a complex series of pre-programmed responses that shift energy to essential maintenance and repair activities, while temporarily shutting down growth and reproduction. The most notable examples of these resilience mechanisms include the UPR and the ISR, likely designed to cope with periods of low nutrient availability or acute and extreme toxic exposures (Fig. 1). The impairment of energy metabolism and the excess production of reactive oxygen species further activate resilience mechanisms but are also confounding factors because resilience requires extra energy.
Although the various stress responses exhibit notable overlap, future research should offer a clearer picture of the specific and unique responses elicited according to the type, intensity, or duration of stress. Future studies should address a number of questions at the intersection of stress and aging. For example, does intermittent stress sensitize cells to senescence? Do aged cells become deficient in mounting an adaptive response, and does this result in increased apoptosis or persistence of damage? Because senescent cells accumulate with aging and are resistant to apoptosis, does the nature of senescence change with successive rounds of stress? In addition, although we know many molecular details of the UPR and ISR, it is unclear how other components of the stress response, such as stress granules and ribonucleoprotein condensates, fit into the canonical responses.
Evidence is accumulating that stress responses encompass mechanisms of maintenance and potentiation that may direct to chronic states. For example, severe ER stress can cause senescence that can induce further senescence through paracrine mechanisms and eventually chronic inflammation at the organismal level. This phenomenon may be a cause of “inflammaging,” a state of chronic inflammation in tissues observed in older individuals and associated with many degenerative diseases of aging72,73. Modulation of stress responses to a level that is beneficial but does not self-perpetuate to the point of being damaging may serve as a strategy to treat inflammaging.
Finally, there is the question of whether the gradual breakdown of resilience responses over time causes the functional manifestation of aging and ultimately frailty and death (Fig. 3). To address this question, it will be important to develop experimental models that can probe the effects of intermittent stress with full control over variables such as intensity, duration, and frequency. These models would be invaluable for testing if there are specific thresholds of stress beyond which the damage is no longer fully reversible, and whether these thresholds change with aging. For instance, while damaged lysosomes can be repaired, whether the repair capacity is reduced in older cells is not known but would be expected given that lysosome membrane permeability increases with aging. An atlas of changes that occur with aging in different autophagy pathways would also be of great value to reveal formal connections between these systems and the aging process. Given the role of IRE1α in regulating genome damage and cellular senescence, two hallmarks of aging, participation of the ISR seems likely. Moreover, impaired UPR or ISR is expected in aging more broadly, considering the prominent role of protein aggregation in age-related neurodegenerative pathologies such as seen in AD. The identification of the hallmarks of aging was a “flywheel” in aging research74, but it mostly focused on the hardware mechanisms of resilience. Understanding the software that regulates the hallmarks in different stressful situations will add invaluable dimensions to research in gerosciences.
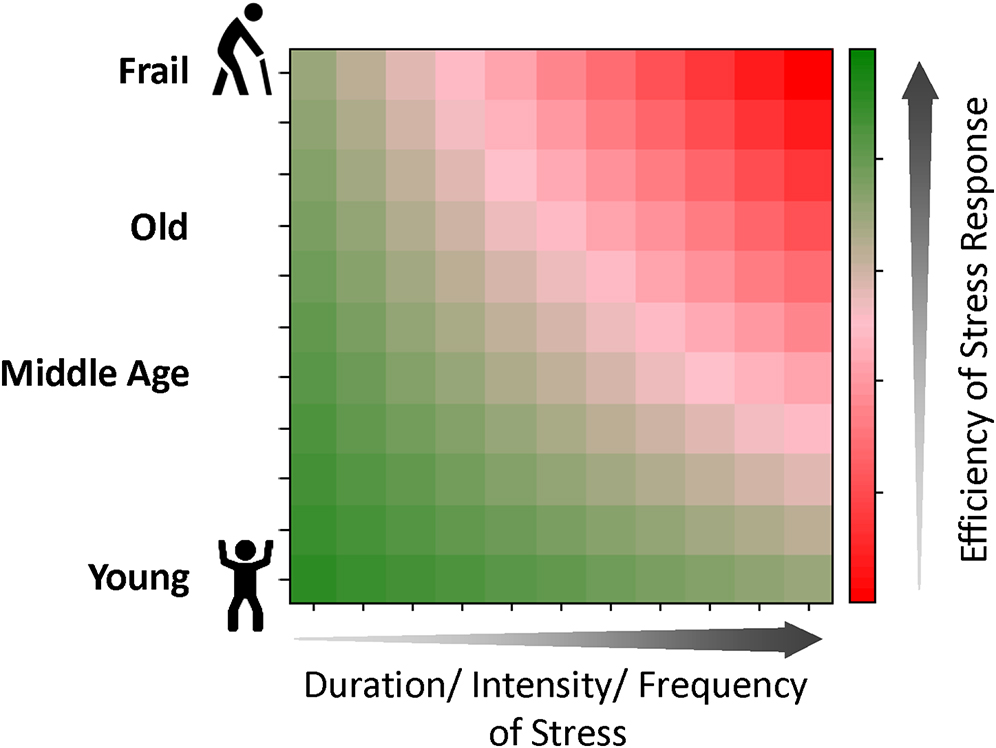
Long duration and high intensity or frequency of stress can exhaust the finite capacity of cells, and correspondingly, the organism’s ability to respond and restore homeostasis. In old age, this ability can be further reduced, leading to an even lower efficiency of the stress response and eventual frailty, a state of biological vulnerability to even mild stressors and limited capacity for recovery.
Acknowledgments
This symposium was supported by the National Institute on Aging (NIA) Intramural Research Program (IRP) and National Institutes of Health (NIH).