Research Paper
Early and Late Chaperone Intervention Therapy Boosts XBP1s and ADAM10, Restores Proteostasis, and Rescues Learning in Alzheimer’s Disease Mice
Authors
Jennifer M. Hafycz,1 Ewa Strus,1 Kamalini Sengupta,1 Nirinjini Naidoo,1,2,*
1Division of Sleep Medicine, Department of Medicine, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
2Chronobiology and Sleep Institute, Perelman School of Medicine, University of Pennsylvania, Philadelphia, PA, USA
*Corresponding author: naidoo@pennmedicine.upenn.edu
DOI:https://doi.org/10.59368/agingbio.20230017
Full Article | PDF | Supplementary
Abstract
Alzheimer’s disease (AD) is a debilitating neurodegenerative disorder that is pervasive among the aging population. Two distinct phenotypes of AD are deficits in cognition and proteostasis, including chronic activation of the unfolded protein response (UPR) and aberrant Aβ production. It is unknown if restoring proteostasis by reducing chronic and aberrant UPR activation in AD can improve pathology and cognition. Here, we present data using an amyloid precursor protein knock-in mouse model of AD and several protein chaperone supplementation paradigms, including a late-stage intervention. We show that supplementing protein chaperones systemically and locally in the hippocampus reduces PERK signaling and increases XBP1s, which is associated with increased ADAM10 and decreased Aβ42. Importantly, chaperone treatment improves cognition, which is correlated with increased CREB phosphorylation and BDNF. Together, these data suggest that chaperone treatment restores proteostasis in a mouse model of AD and that this restoration is associated with improved cognition and reduced pathology.
Introduction
Alzheimer’s disease (AD) is a prevalent neurodegenerative disease and is the leading cause of dementia in older adults1–3. Despite its widespread nature, the causes of AD pathology are not fully understood, and research to uncover these mechanisms and therapies is vital. Indeed, AD is oftentimes difficult to diagnose early on and is typically identified following cognitive impairments, which occur after significant disease progression3–5. The pathogenesis of many neurodegenerative diseases includes protein dyshomeostasis and corresponding protein aggregation6. In AD, these aggregations are commonly seen as tau tangles or amyloid-β aggregations, known as plaques, which are made up of Aβ-42 fibrils7–9. Aβ-42 is generated via amyloidogenic cleavage of amyloid precursor protein (APP) by β-site APP cleaving enzyme 1 (BACE1)10,11. Under healthy conditions, APP is an important synaptic stabilizing protein and is cleaved by an α-secretase, a disintegrin and metalloproteinase 10 (ADAM10), in the non-amyloidogenic pathway10–12. ADAM10 is reduced in AD, which is thought to contribute to the pathogenesis of the disease10,11,13. While the exact detrimental effects of these protein aggregations are unclear, it is thought that Aβ-42 and these aberrant aggregates alter neuronal signaling and thus contribute to cognitive decline14,15.
The maintenance of protein homeostasis, or proteostasis, is critical for the function and survival of the cell. The protein aggregations seen in AD are indicative of an imbalance in proteostasis and an increase in endoplasmic reticulum (ER) stress6,16. ER stress occurs when proteins misfold and aggregate in the ER lumen17–20. ER stress in healthy organisms activates the unfolded protein response (UPR), a protein quality control and signal transduction pathway21. The UPR restores proteostasis by alleviating the protein folding load on the cell through the activation of three distinct ER stress sensors: PERK, IRE1, and ATF621–24. Briefly, PERK activation attenuates global protein translation, apart from a few key targets, including activating transcription factor 4 (ATF4)21. IRE1 activation leads to increased spliced x-box binding protein 1 mRNA and the generation of the transcription factor X-Box Binding Protein 1 (XBP1s), which promotes the synthesis of the chaperone protein, BiP25, while ATF6 activation also promotes chaperone synthesis21. Collectively, these pathways work to resolve ER stress and restore proteostasis.
Importantly, age is a major risk factor for the development of AD, and there is a plethora of evidence that demonstrates increased UPR activity in AD brains, both in postmortem human tissue as well as in several animal models of AD and other neurodegenerative diseases6,16,26–28. Under healthy aging conditions, the mechanisms that maintain proteostasis are less efficient and can even become maladaptive24,29–31. Specifically, with age, there is increased ER stress, and the UPR is more chronically activated23,24,29,30,32,33. Chronic UPR activity promotes pro-apoptotic signaling and cell death via prolonged PERK signaling and subsequent CHOP activation23,33. With age, there is also a reduction in the endogenous ER protein chaperone BiP, which is crucial to prevent protein misfolding21,24,29,31,34. Furthermore, chronically inhibited protein translation via PERK activation has many consequences, including memory deficits35–39. Interestingly, the UPR can modulate ADAM10 levels, as XBP1s, downstream of IRE1, acts as a transcription factor to promote ADAM1040,41. However, it remains unclear if UPR activation is a symptom of AD or if chronic maladaptive UPR activation is causal to the progression of the disease. Furthermore, it is unknown if modulating the UPR can alter XBP1s levels, and thus ADAM10, to improve AD pathology.
We have shown previously, in both drosophila and mouse models of aging, that supplementing chaperone levels with a small chemical chaperone, 4-phenylbutyrate (PBA), reduces chronic UPR activity in the brain29,30. Here, we wanted to determine if UPR activity played a similar role in cognition and pathology in an APP knock-in (KI) mouse model of AD. We used the APPNL-G-F model, developed to include the Swedish, Iberian, and Arctic mutations, which uses the endogenous mouse APP promoter to drive the expression of APP42. We employed three distinct groups of these APPNL-G-F KI mice and their APPWT wildtype (WT) littermates to determine if intervening at an early age or at a later age, when the disease has progressed, can improve behavior and pathology.
We report that chaperone treatment improved cognitive performance with both early and late-stage intervention in the APPNL-G-F KI mice. Chaperone treatment also led to improved proteostasis, with a diminution of ER stress in the hippocampus, an increase in ADAM10 and CREB activation, and a decrease in Aβ-42 in APPNL-G-F KI mice. Promisingly, hippocampal BiP overexpression recapitulated these results. Altogether, our data demonstrate that disrupted proteostasis and chronic UPR activity play a key role in AD and that supplementing chaperone levels is sufficient to restore both proteostasis and cognition during early and late-stage intervention paradigms. Together, this work could inform the development of novel therapeutics to help treat this debilitating disease.
Materials and Methods
Mice
Animal experiments were conducted in accordance with the guidelines of the University of Pennsylvania Institutional Animal Care and Use Committee. Mice were maintained as previously described24,89. Mice were housed at 23°C on a 12:12-hour light/dark cycle and had ad libitum access to food and water. APPNL-G-F KI and APPWT WT mice were used for these experiments42. Two groups of mice were used for PBA treatment studies, and a third group was used for AAV-BiP overexpression. Animal numbers for each genotype, sex, and treatment condition used for spatial object recognition (SOR) cognitive testing can be found in Supplemental Table 5.
Drug administration
Sodium PBA (Cayman Chemical, Ann Arbor, MI) was administered twice weekly via intraperitoneal (IP) injections at a dose of 40 mg/kg and in drinking water (0.008% PBA in 1% sucrose) as previously described30. Vehicle-treated mice received IP sterile saline injections twice weekly and a 1% sucrose solution in drinking water. Treatment began at weaning (4−6 weeks) or at 10−12 months of age (late intervention) and continued for 10−12 weeks, followed by cognitive behavioral testing and tissue harvest as previously described30.
Stereotaxic injection surgery
Stereotaxic hippocampal injections were performed to deliver a BiP viral overexpression vector (Vector Biolabs; AAV5-CamKIIa-GRP78; titer 4.8 × 1012 GC/ml) and an mCherry control vector (Addgene; pAAV5-CaMKIIa-mCherry; titer 2.3 × 10−13 GC/ml) into the hippocampi of young and aged mice, as previously described30. Four burr holes were drilled for bilateral hippocampal injections, with coordinates as follows: AP ± 1.8 mm, ML ± 0.8, ±1.8 mm, DV −1.7, and −1.9 mm. Using a 1 μL Hamilton syringe, 50 nanoliters of virus were injected per burr hole. Behavioral testing occurred four weeks after surgery, followed by tissue harvest as previously described30.
SOR (Spatial Object Recognition) test
The SOR test is a well-established hippocampal-dependent spatial memory test30,46,47. SOR was carried out as previously described, with a training phase followed by a testing phase30. Discrimination index calculations were performed as previously described as a measure for how well the mice distinguished between the moved and unmoved object48.
Y-maze spontaneous alternation test
The Y-Maze test was performed as previously described49. Briefly, a single mouse was placed in the center of the apparatus and allowed to move freely through the maze for 5 min. Each individual arm entry and the order in which the entries occurred were recorded. After testing, the number of alternations (three separate arm sequential arm entries) was counted and presented as a percentage.
Immunohistochemical assays
Postfixed half-brain coronal sections were sliced at 40 μm using a cryostat as previously described30,90. Every other section was placed in 24-well plates containing cryoprotectant for free floating immunohistochemistry staining and stored at −20 °C, as previously described24,91. For all markers, we compared n = 4–8 in each of the four groups.
Immunofluorescence (IF)
IF staining was conducted as previously described, with an n = 4–8 animals/group/assay30,31. Antibodies used are listed in SI.
Quantitative analysis of IF images
Confocal images were acquired as previously described30,92 using a Leica SP5/AOBS microscope. Confocal laser intensities, nm range, detector gain, exposure time, amplifier offset, and depth of the focal plane within sections per antigen target were standardized across the compared sections. Confocal images were quantified as previously described93. Briefly, three to four sections were imaged per animal (n = 4–5 mice/group). Using ImageJ software, the images were converted to an 8-bit grayscale with a detection threshold standardized across all images to detect percent areas. The percentage area covered within the target region was measured, and the average percent areas for each mouse were analyzed.
Western-blot staining
Frozen brain tissue was prepared for western-blot assays as previously described24,30,91. Homogenized tissue samples (20 μg protein) were separated by SDS-PAGE, and protein bands were imaged and quantified via infrared imaging on an Odyssey scanner (LiCor). For all markers, n = 3–8 samples were compared. Primary and secondary antibodies used are listed in SI.
Statistical analyses
Data are presented as the average ± standard error of the mean of sample size n. Statistical analyses were performed in PRISM (GraphPad Software, La Jolla, CA). A two-way ANOVA was used to determine interaction effects, with Tukey post hoc corrections for multiple comparisons. p < 0.05 was the threshold for determining statistical significance. In some instances, as interaction tests are typically underpowered, regardless of the significance of the ANOVA term, we subsequently performed specific between-group comparisons defined by APPNL-G-F genotype and treatment. We evaluated whether outcome measures differed between genotype and treatment using two-tailed T-tests (see supplement). Together, this set of analyses provides a more comprehensive evaluation of the APPNL-G-F genotype and treatment group relationships.
Results
Early PBA treatment improves proteostasis in the hippocampus of APPNL-G-F KI mice
Given the prevalence of UPR activity in brain tissues from AD patients and animal models6,16,28 and the fact that aberrant UPR activity, present in aging, is coupled with diminished BiP levels24,29,30, we first wanted to establish if BiP is inherently reduced in APPNL-G-F KI mice. Since the APPNL-G-F mutations are constitutive, we intervened with IP PBA or saline injections starting from weaning. Following 10 weeks of treatment and behavioral testing, we used IF to probe for BiP and subsequent markers of UPR activity.
Early PBA treatment restores BiP levels in the APPNL-G-F KI mice
We found that BiP levels are reduced in the hippocampus of saline-treated APPNL-G-F KI mice, most notably in the CA3 region (p < 0.001; Fig. 1A), but also in the CA1 and dentate gyrus (p < 0.01 and p < 0.05, respectively; Supplemental Fig. 1A,B; Supplemental Table 1) compared to saline-treated WT littermate controls. Following PBA treatment, the APPNL-G-F KI mice displayed restored levels of BiP in the CA3 (p < 0.01; Fig. 1A; Supplemental Table 1), as well as in the dentate gyrus (p = 0.05; Supplemental Fig. 1A,B; Supplemental Table 1), indicative of an increase in endogenous chaperone synthesis with PBA treatment.
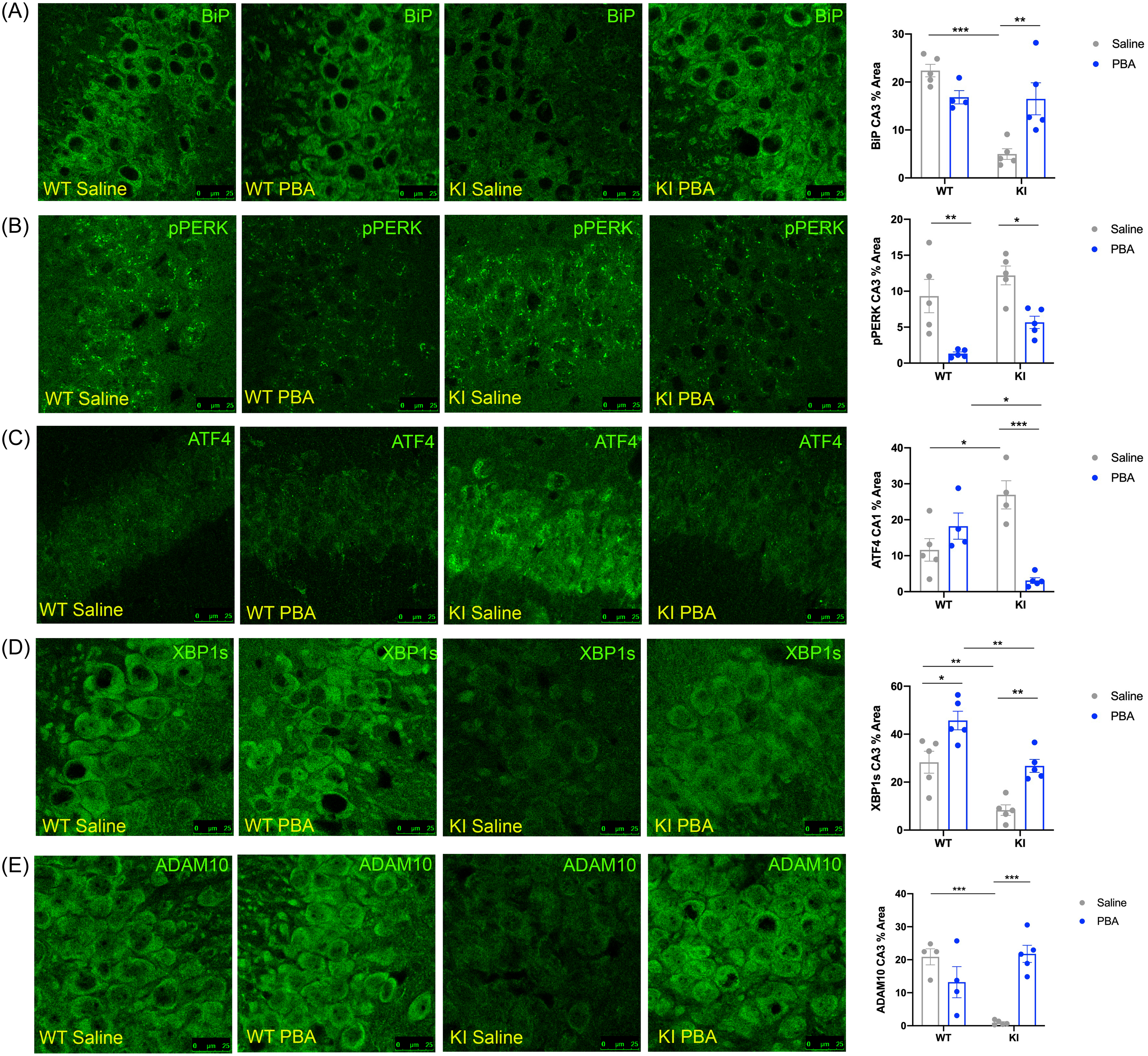
Confocal images of the hippocampus across groups. (A) BiP in the CA3. (B) p-PERK in the CA3. (C) ATF4 in the CA1. (D) XBP1s in the CA3. (E) ADAM10 in the CA3. Data quantified and presented as mean ± SE percent area of BiP, p-PERK, ATF4, XBP1s, and ADAM10 within hippocampal sections (n = 4–5 animals per group; two-way ANOVA with Tukey post hoc correction for multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001).
Early PBA treatment reduces PERK signaling
Using IF to assess PERK activation, we found no difference in p-PERK levels in the CA3 region of the hippocampus in APPNL-G-F KI mice when compared to APPWT WT mice under saline-treated conditions (p > 0.4; Fig. 1B; Supplemental Table 1). Both APPNL-G-F KI and APPWT WT PBA-treated mice showed a decrease in PERK phosphorylation in the CA3 compared to saline-treated mice (p < 0.05 and p < 0.01, respectively; Fig. 1B; Supplemental Table 1), demonstrating that PBA treatment can reduce baseline levels of UPR activity independent of genotype.
Next, we assessed levels of activating transcription factor 4 (ATF4), which is a transcription factor that bypasses the translation block induced by p-PERK phosphorylation of eIF2α. APPNL-G-F KI mice displayed more ATF4 staining in the CA1 than APPWT WT littermates (p < 0.05; Fig. 1C; Supplemental Table 1). APPNL-G-F KI PBA-treated mice had less ATF4 in the CA1 relative to saline-treated APPNL-G-F KI mice (p < 0.001; Fig. 1C; Supplemental Table 1). This suggests that PBA treatment from weaning reduces ATF4, which is downstream of the PERK-peIF2α translation block, in the hippocampi of APPNL-G-F KI mice.
Early PBA treatment in APPNL-G-F KI mice increases XBP1s and ADAM10
In addition to PERK signaling, we also examined activation of IRE1, which leads to XBP1s synthesis. XBP1s contributes to an adaptive pro-survival UPR response, as XBP1s promotes chaperone synthesis21,25. We hypothesized that APPNL-G-F KI mice exhibit more chronic, maladaptive UPR activity and thus display a diminished adaptive response. Using IF, we found that saline-treated APPNL-G-F KI mice had less XBP1s relative to WT littermates in the CA3 of the hippocampus (p < 0.01; Fig. 1D; Supplemental Table 1), as well as in the dentate gyrus (p < 0.05; Supplemental Fig. 1D). PBA treatment from weaning increased XBP1s in both APPNL-G-F KI and APPWT WT mice in the CA3 (p < 0.01, p < 0.05, respectively; Fig. 1D; Supplemental Table 1), as well as in the CA1 of APPNL-G-F KI and APPWT WT (p < 0.01 and p < 0.001, respectively; Supplemental Fig. 1C) and the dentate gyrus (p < 0.05 and p < 0.05, respectively; Supplemental Fig. 1D), altogether demonstrating that an early intervention with PBA treatment increases an adaptive UPR response via promoting XBP1s.
Importantly, XBP1s regulates ADAM10, which is an α-secretase that promotes non-amyloidogenic cleavage of APP10,40. We probed for ADAM10 to determine if the changes observed in UPR activity are linked to APP processing. We observed less ADAM10 in the CA3 region of the hippocampus in saline-treated APPNL-G-F KI mice compared to saline-treated APPWT WT mice (p < 0.001; Fig. 1E; Supplemental Table 1). With PBA treatment, the APPNL-G-F KI mice displayed increased ADAM10 levels compared to saline-treated APPNL-G-F KI mice (p < 0.001; Fig. 1E; Supplemental Table 1). These data indicate that chaperone treatment could affect how APP is processed by altering the amount of ADAM10 downstream of XBP1s.
As PBA also acts as an HDAC inhibitor, we examined the acetylation of histones H3 and H4 and found that PBA treatment did not alter the acetylation of these histones (Supplemental Fig. 1E).
PBA treatment at an early age improves learning in APPNL-G-F KI mice
Memory deficits are a central phenotype of AD3,4,43. Many studies have shown previously that memory deficits are linked to increased ER stress30,44,45. To probe cognitive deficits in the APPNL-G-F KI mice and to establish the effect of PBA treatment on cognition, both male and female APPNL-G-F KI and APPWT WT mice were subjected to the SOR cognitive task and the Y-maze task (Fig. 2A). Learning was quantified in the SOR test by calculating a discrimination index between the moved and unmoved objects30,46–48, while spatial working memory in the Y-maze test was measured by calculating percent alternations30,49.

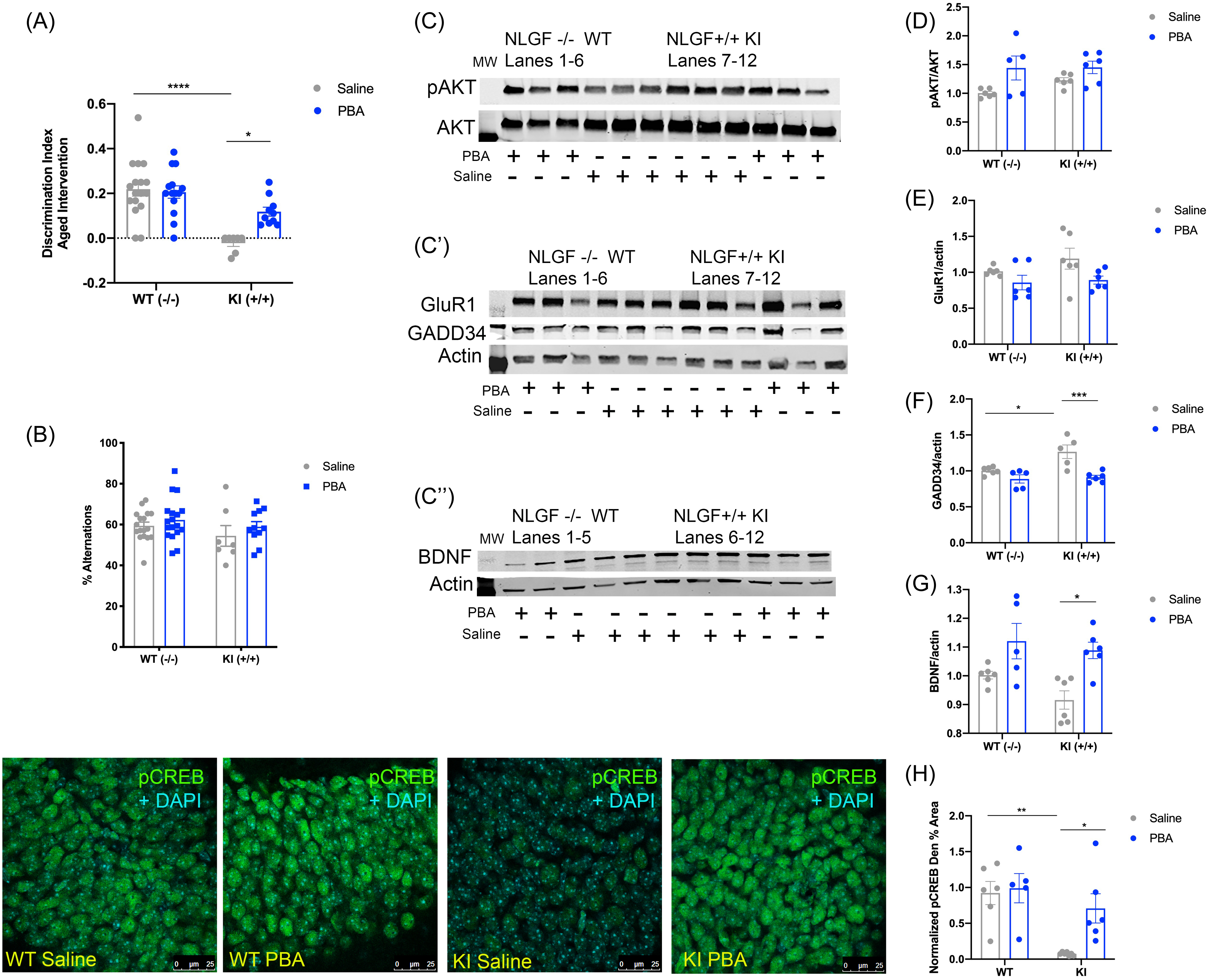
(A) Schematic of SOR and Y-maze tests. (B) Discrimination index of the SOR test. (C) Percent alternations from the Y-maze test. (D–D″) Representative western-blot images; each panel is a different animal. Membranes are cut along the molecular weight (MW) markers following blocking to use the two-channel Odyssey system and varying molecular weights for the proteins of interest to probe for multiple markers within one membrane: (D) pAKT and AKT, ∼65 kDa; (D′) GluR1 ∼110 kDa, GADD34 ∼100 kDa, Actin ∼42 kDa; and (D″) BDNF dimer ∼28 kDa, Actin ∼42 kDa. (E) Quantification of western-blot analysis for pAKT/AKT. (F) Quantification of western-blot analysis for GADD34/actin. (G) Quantification of western-blot analysis for GluR1/actin. (H) Quantification of western-blot analysis for BDNF/actin. Bottom: Confocal images of the hippocampal dentate gyrus across groups. (I) Mean ± SE percent area of pCREB in the dentate gyrus; n = 4–6 animals per group for western-blot and immunohistochemical assays. (All data presented as mean ± SE; all quantifications analyzed via two-way ANOVA with Tukey post hoc correction for multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001).
Early PBA treatment improves SOR test performance in APPNL-G-F KI mice
We found that APPWT WT mice were able to discriminate between the moved and unmoved objects in the SOR test regardless of treatment. APPNL-G-F KI mice performed poorly in this test compared to saline-treated WT littermates, with a clear inability to discriminate between the two objects (p < 0.001; Fig. 2B; Supplemental Table 4). With PBA treatment, APPNL-G-F KI mice showed improved performance, being able to distinguish between the moved and unmoved objects (p < 0.001; Fig. 2B; Supplemental Table 4; Supplemental Fig. 4A). Male and female mice performed similarly within genotype in response to saline and PBA treatment (Supplemental Fig. 5A,B). However, there were no differences observed in performance in the Y-maze test across treatment or genotype (Fig. 2C). Together, these data suggest that PBA treatment improves spatial learning in the APPNL-G-F KI mice.
Early PBA treatment increases phosphorylation of CREB and AKT in the hippocampus of APPNL-G-F KI mice
We have shown that chaperone treatment correlates with increased CREB phosphorylation in the dentate gyrus of the hippocampus in aged mice30. To determine if a similar mechanism is at work in the APPNL-G-F KI model, we examined CREB activation. Male and female mice were pooled together for all IF assays, as SOR performance was not different between males and females. n = 4–5 mice per group were used for IF assays to best correlate changes in SOR performance with changes at the cellular level.
Saline-treated APPNL-G-F KI mice displayed reduced levels of p-CREB in the dentate gyrus compared to saline-treated APPWT WT mice (p < 0.01; Fig. 2; Supplemental Table 1). Following PBA treatment, APPNL-G-F KI mice showed an increase in phosphorylated CREB relative to saline-treated APPNL-G-F KI mice (p < 0.05; Fig. 2; Supplemental Table 1).
Previous work has shown that the activation of the CREB kinase, AKT, can be altered by the UPR through GADD34, which is downstream of p-PERK and ATF430,50,51. We next examined APPNL-G-F KI and WT tissue for p-AKT and GADD34 via western-blot assays. We observed less p-AKT in the hippocampus of saline-treated APPNL-G-F KI mice compared to saline-treated APPWT WT mice (p < 0.05; Fig. 2D,E). Following PBA treatment, APPNL-G-F KI mice displayed increased levels of p-AKT compared to saline-treated APPNL-G-F KI mice (p < 0.05; Fig. 2D,E). However, we observed no changes in GADD34 across the groups (Fig. 2D′,F).
CREB activation is regulated by synaptic activity; specifically, AMPA signaling is known to lead to CREB activation, and AMPA levels are known to be altered in AD52–55. Thus, we quantified the AMPA subunit GluR1 via western-blot assays in the hippocampus of saline- and PBA-treated APPNL-G-F KI and WT mice. We observed no changes in GluR1 levels in any groups, regardless of genotype or treatment (Fig. 2D′,G). To further establish if changes in p-CREB levels are related to changes in cognition, we used western-blot assays to probe for BDNF, a growth factor downstream of CREB well known for its role in memory formation56–58. Western analyses did not reveal an obvious deficit in BDNF in the APPNL-G-F KI saline-treated mice relative to APPWT WT saline-treated mice (Fig. 2D″,H). While PBA treatment did indicate a trend toward increased BDNF levels in the APPNL-G-F KI mice compared to saline-treated APPNL-G-F KI mice, it was not significant (p = 0.06; Fig. 2D″,H). Together, these data suggest that chaperone treatment could improve cognition by increasing p-CREB signaling and potentially BDNF in the APPNL-G-F KI mice via modulating p-AKT activation.
Late-stage PBA intervention reduces maladaptive UPR activity and improves learning in older APPNL-G-F KI mice
As AD is generally not diagnosed until after serious cognitive deficits have manifested4,5, we carried out a late-stage PBA intervention paradigm on a second group of APPNL-G-F KI and APPWT WT mice starting at 10–12 months of age and continuing for 10–12 weeks.
Late-stage PBA intervention increased BiP and reduced CHOP levels
Probing for BiP, we found that these aged saline-treated APPNL-G-F KI mice also exhibited reduced levels of BiP in the CA3 and dentate gyrus of the hippocampus compared to saline-treated APPWT WT mice (p < 0.001 and p < 0.05, respectively; Fig. 3A; Supplemental Fig. 3A; Supplemental Table 3). BiP levels were restored with PBA treatment in the APPNL-G-F KI mice in the CA3 and dentate (p < 0.01 and p < 0.05, respectively; Fig. 4; Supplemental Fig. 3A; Supplemental Table 3). PERK phosphorylation and ATF4 levels were not altered between the aged APPNL-G-F KI and APPWT WT mice regardless of treatment (IF images not shown; Supplemental Table 3), suggesting that delaying chaperone treatment is not sufficient to combat exacerbated PERK phosphorylation. We also probed for CHOP in the hippocampus of these late-stage PBA intervention mice. CHOP is a transcriptional target of ATF4 and is known to induce pro-apoptotic signaling21,22,59. We found that saline-treated APPNL-G-F KI mice had more CHOP in the CA3 region of the hippocampus than saline-treated APPWT WT mice (p < 0.001; Fig. 3B; Supplemental Table 3). PBA-treated APPNL-G-F KI mice displayed reduced CHOP in the CA3 relative to saline-treated APPNL-G-F KI mice (p < 0.01; Fig. 3B; Supplemental Table 3). This indicates that while upstream PERK activity is not altered with PBA treatment, downstream pro-apoptotic signaling via the PERK-ATF4-CHOP pathway is reduced with PBA in APPNL-G-F KI mice.
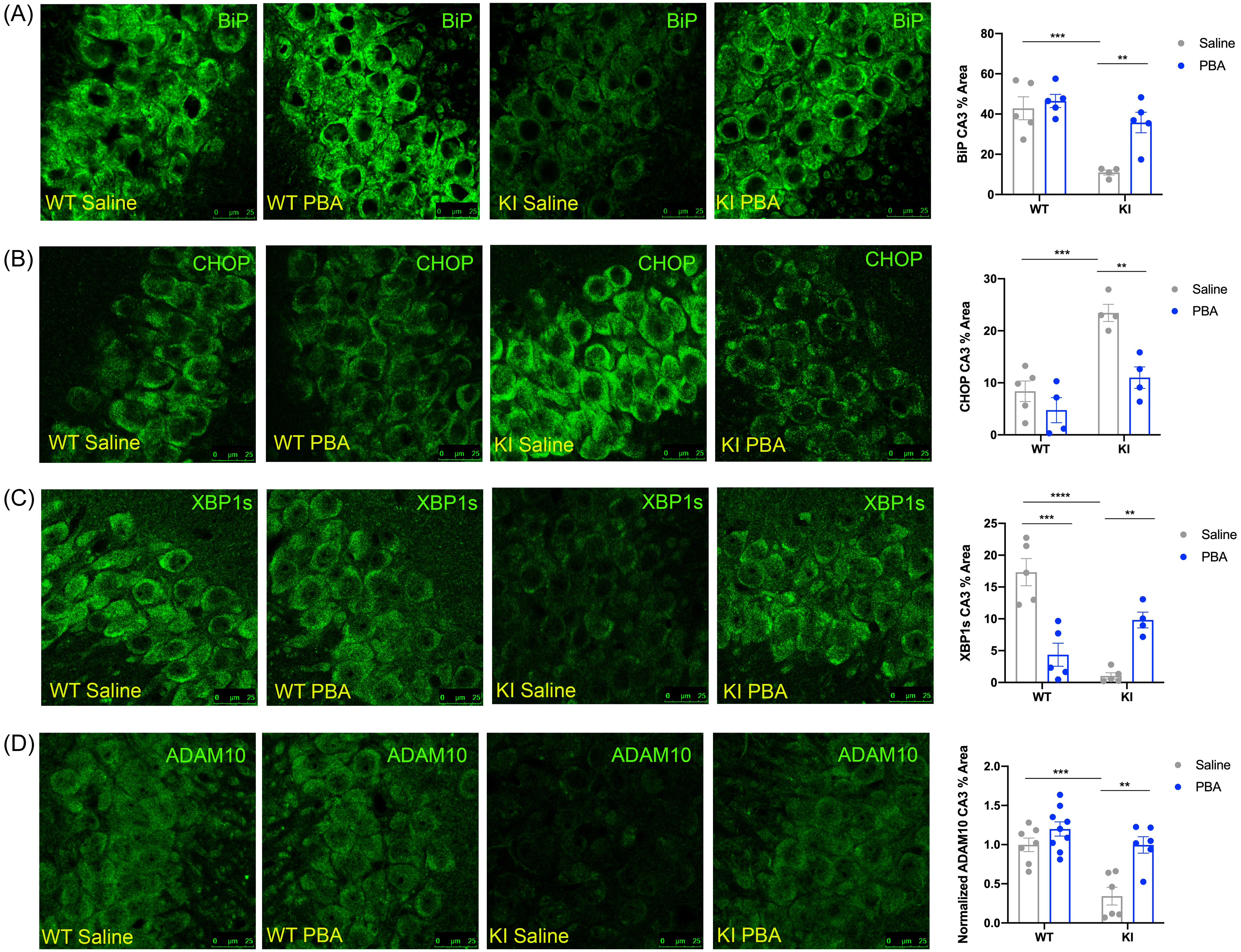
Representative confocal images of the hippocampus across groups. (A) BiP immunofluorescent images in the CA3. (B) CHOP immunofluorescent images in the CA3. (C) XBP1s immunofluorescent images in the CA3. (D) ADAM10 immunofluorescent images in the CA3. (Data presented as mean ± SE percent area of BiP, CHOP, XBP1s, and ADAM10 immunofluorescence (IF) within hippocampal sections; n = 4–9 animals per group; two-way ANOVA with Tukey post hoc correction for multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
(A) Discrimination index of the SOR test. (B) Percent alternations in the Y-maze test. (C–C″) Representative western-blot images. Each panel is a different animal. Membranes are cut along the MW markers following blocking to use the two-channel Odyssey system and varying molecular weights for the proteins of interest to probe for multiple markers within one membrane: (C) pAKT and AKT, ∼65 kDa; (C′) GluR1 ∼110 kDa, GADD34 ∼100 kDa, Actin ∼42 kDa; and (C″) BDNF dimer ∼28 kDa, Actin ∼42 kDa. (D) Quantification of western-blot analysis for pAKT/AKT. (E) Quantification of western-blot analysis for GADD34/actin. (F) Quantification of western-blot analysis for GluR1/actin. (G) Quantification of western-blot analysis for BDNF/actin. Bottom Right: Confocal images of the hippocampal dentate gyrus across groups. (H) Quantification of pCREB immunofluorescence normalized to APPWT WT mice, mean ± SE percent area within hippocampal dentate gyrus sections. n = 5–6 animals per group for western-blot and immunohistochemical assays. (All data presented as mean ± SE; all quantifications analyzed via two-way ANOVA with Tukey post hoc correction for multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001).
Late-stage PBA treatment increased XBP1s and ADAM10 in APPNL-G-F KI mice
In addition, we examined XBP1s levels and found that saline-treated APPNL-G-F KI mice displayed less XBP1s in the CA3 of the hippocampus relative to saline-treated APPWT WT mice (p < 0.0001; Fig. 3C; Supplemental Table 3), as well as in the CA1 (p < 0.01; Supplemental Fig. 2). With PBA treatment, APPNL-G-F KI mice exhibited restored levels of XBP1s in the CA3 and CA1 compared to saline-treated APPNL-G-F KI mice (p < 0.01 and p < 0.01, respectively; Fig. 3C; Supplemental Fig. 2; Supplemental Table 3). Together, these data demonstrate that even a late-stage chaperone intervention can reduce chronic maladaptive UPR signaling and restore adaptive UPR functions.
To determine if APP processing could be altered with PBA treatment, we probed for ADAM10 in these late-stage intervention mice. We found that there was less ADAM10 staining in the CA3 of saline-treated APPNL-G-F KI mice compared to APPWT WT mice (p < 0.001; Fig. 3D; Supplemental Table 3). With PBA treatment, ADAM10 was increased in the CA3 of APPNL-G-F KI mice relative to saline-treated APPNL-G-F KI mice (p < 0.01; Fig. 3D; Supplemental Table 3). This indicates that a late-stage chaperone intervention leads to increased ADAM10 through modulation of the UPR response.
PBA intervention at a later stage improved learning in the APPNL-G-F KI mice
To determine if late-stage chronic PBA treatment can improve learning in the older (12–14 months at the time of testing) APPNL-G-F KI mice following 10 weeks of either saline or PBA injections, all mice were subjected to the SOR test. As before, saline-treated APPNL-G-F KI mice were unable to distinguish between the moved and unmoved objects compared to saline-treated APPWT WT mice (p < 0.0001; Fig. 4A; Supplemental Table 4). With PBA treatment, the APPNL-G-F KI mice were able to distinguish between the objects, indicative of improved learning (p < 0.05; Fig. 4A; Supplemental Fig. 4B; Supplemental Table 4). We observed no differences between males and females in this learning test (Supplemental Fig. 5E,F). We also observed no differences in percent alternations in the Y-maze test across groups (Fig. 4B). Promisingly, overall, this indicates that even a late-stage interventional chaperone treatment can improve learning in the APPNL-G-F KI mice.
Late-stage PBA intervention increased CREB phosphorylation and BDNF levels in the APPNL-G-F KI mice
Next, we measured CREB phosphorylation to determine if the improvement in learning was correlated with increased p-CREB. Consistent with earlier observations, we found that APPNL-G-F KI mice that received only saline injections had less p-CREB in the dentate gyrus than saline-treated APPWT WT mice (p < 0.01; Fig. 4; Supplemental Table 3). With PBA treatment, APPNL-G-F KI mice had increased phosphorylation of CREB compared to saline-treated APPNL-G-F KI mice (p < 0.05; Fig. 4; Supplemental Table 3). We next examined activation of AKT and quantified p-AKT using western blots and found that p-AKT levels were unchanged regardless of genotype or treatment (Fig. 4C,D). We, however, found more hippocampal GADD34 in these late-stage APPNL-G-F KI saline-treated mice when compared to APPWT WT saline-treated mice (p < 0.05; Fig. 4C′,F). PBA treatment led to a reduction in GADD34 in the APPNL-G-F KI mice compared to APPNL-G-F KI saline-treated mice (p < 0.001; Fig. 4C′,F). To determine if AMPA levels were changed with a late-stage treatment paradigm, we also probed for GluR1 via western blot. However, consistent with our other experimental paradigms, we observed no changes in the GluR1 amount across groups. Interestingly, APPNL-G-F KI saline-treated mice did not display less hippocampal BDNF as compared to APPWT WT mice (Fig. 4C″,G), yet late-stage PBA treatment resulted in increased BDNF in the APPNL-G-F KI mice (p < 0.05; Fig. 4C″,G). Collectively, these data suggest that even a late chaperone intervention can improve cognition, with corresponding increases in p-CREB and BDNF.
Late-stage PBA treatment reduces Aβ42 in APPNL-G-F KI mice
One of the key pathological features of AD is amyloid-β plaques that consist of insoluble Aβ fibrils60,61. To determine if PBA treatment improved pathology, we carried out immunostaining using the 6E10 antibody, which labels Aβ plaques.
We did not observe any gross differences in plaque number in the hippocampi of early PBA and saline-treated APPNL-G-F KI mice (Fig. 5A), but we did observe a significant decrease in plaque number in mice that received late-stage treatment (Fig. 5B). As there is evidence that Aβ oligomers, as opposed to plaques, are the more toxic Aβ species60,61, we also probed Aβ42 levels using ELISAs in the late-stage older APPNL-G-F KI mice and found that PBA reduced the concentration of Aβ42 in the hippocampus of these mice compared to saline-treated APPNL-G-F KI mice (Fig. 5C). These results are consistent with the changes we observed in ADAM10 levels with PBA treatment, suggesting that modulating the UPR via PBA treatment can alter APP processing via increasing ADAM10.

Confocal images of the hippocampus across groups. (A) 6E10 immunofluorescent images and plaque number in the CA3 in APPNL-G-F KI mice that received treatment from weaning. (B) 6E10 immunofluorescent images and plaque number in the CA3 in APPNL-G-F KI mice that received late-stage treatment. (C) ELISA data using hippocampal homogenate from late-stage APPNL-G-F KI mice. (Data presented as mean ± SE; n = 4–5 animals per group for 5A and 5C and n = 9–12 animals per group for 5B; T-test, *p < 0.05; **p < 0.01).
Binding Immunoglobulin Protein (BiP) overexpression in the hippocampus of APPNL-G-F KI mice improves proteostasis and cognition
Having observed the effect of systemic chaperone administration on learning and UPR molecular markers, we wanted to determine whether restoring chaperone levels in the hippocampus specifically would be sufficient to restore cognitive function and proteostasis in the APPNL-G-F KI mice. Using bilateral hippocampal stereotaxic surgical microinjections of an AAV-CaMKII-BiP, we overexpressed the endogenous chaperone, BiP, in the hippocampus of adult (6-month-old) APPNL-G-F KI and WT littermate mice. An AAV-CaMKII-mCherry vector was used for control microinjections.
BiP overexpression was confirmed with IF staining and western blotting (Fig. 6A; Supplemental Fig. 2; Supplemental Table 2). BiP was markedly increased in the CA3 of APPNL-G-F KI mice (p < 0.001; Fig. 6A) and in the dentate gyrus of APPNL-G-F KI mice (p < 0.01; Supplemental Fig. 2; Supplemental Table 2). Control injections were also confirmed by mCherry staining in the CA3 and dentate gyrus (Fig. 6A; Supplemental Fig. 2).
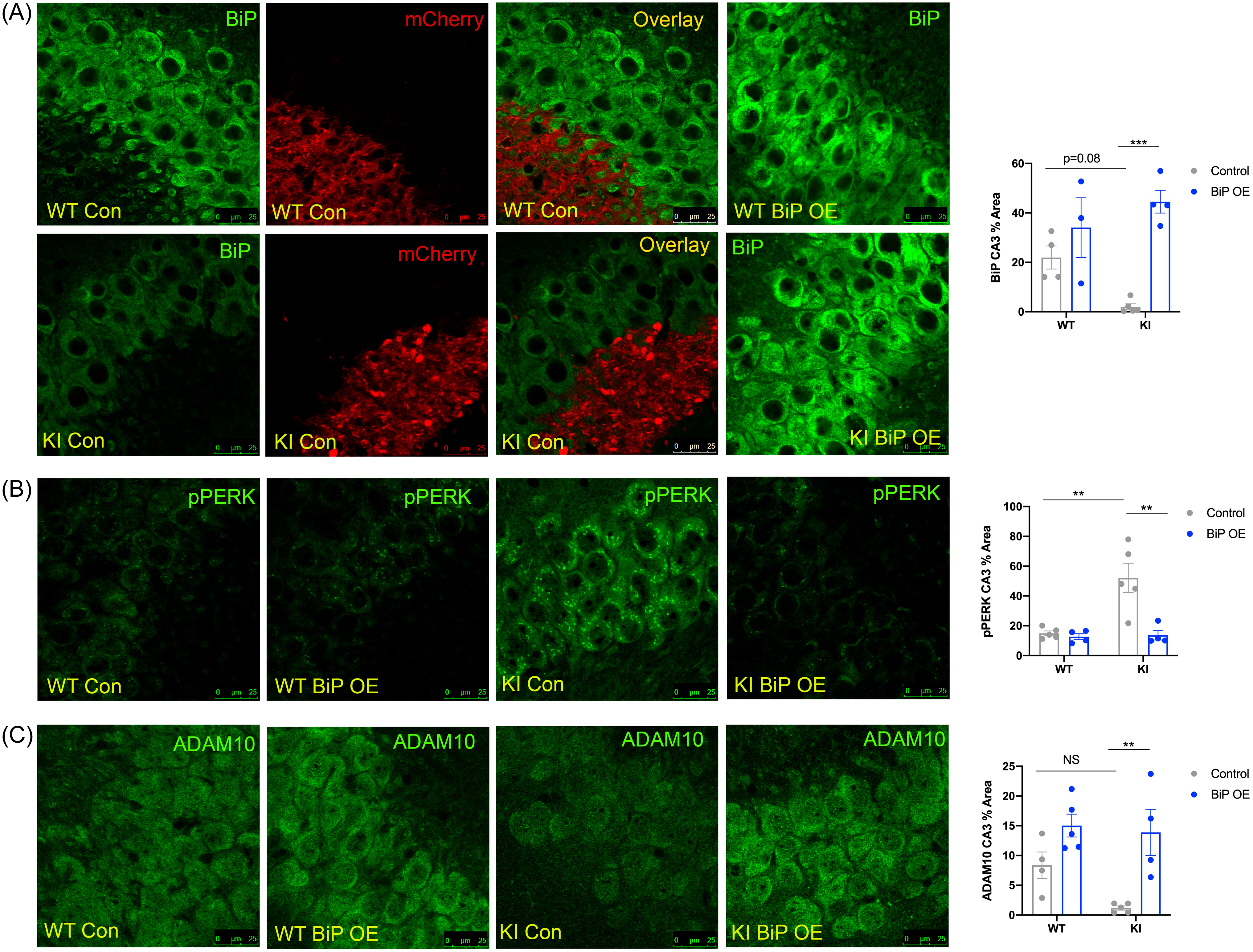
Confocal images of the hippocampal CA3 across groups. (A) BiP staining and merged images shown for mCherry-tagged (red) control virus-injected mice. (B) p-PERK. (C) ADAM10. Mean ± SE percent area within hippocampal sections (n = 3–5 animals per group; two-way ANOVA with Tukey post hoc correction for multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001).
Hippocampal BiP overexpression reduces PERK phosphorylation and increases ADAM10 in the APPNL-G-F KI mice
Following confirmation of viral targeting and BiP overexpression, we probed for markers of UPR activation. APPNL-G-F KI mice that received control injections displayed more p-PERK staining in the CA3 than control-injected APPWT WT mice (p < 0.01; Fig. 6B; Supplemental Table 2). With BiP overexpression, APPNL-G-F KI mice exhibited reduced p-PERK levels (p < 0.01; Fig. 6B; Supplemental Table 2), indicating that overexpressing BiP directly into the hippocampus is sufficient to reduce PERK phosphorylation.
To determine if BiP overexpression was sufficient to promote non-amyloidogenic APP processing, we probed for ADAM10. With BiP overexpression, APPNL-G-F KI mice showed increased ADAM10 compared to control-injected APPNL-G-F KI mice (p < 0.01; Fig. 6C; Supplemental Table 2).
Overexpressing BiP in the hippocampus of APPNL-G-F KI mice improves learning
Next, we wanted to confirm if hippocampal BiP overexpression was sufficient to improve learning. We tested learning in APPNL-G-F KI and APPWT WT mice following BiP and control injections using the SOR and Y-maze tests. We found that the APPWT WT mice, regardless of injection, were able to successfully distinguish between the moved and unmoved objects in the SOR test (Fig. 7A; Supplemental Table 4). However, APPNL-G-F KI control-injected mice were unable to differentiate between the objects compared to control-injected APPWT WT mice (p < 0.001; Fig. 7A; Supplemental Table 4). APPNL-G-F KI that received hippocampal BiP overexpression injections showed improved learning in the SOR test and were able to discriminate between the objects compared to control-injected APPNL-G-F KI mice (p < 0.001; Fig. 7A; Supplemental Table 4). Again, there were no differences in how males and females performed in the test (Supplemental Fig. 5C,D). There were also no differences across groups in percent alternations in the Y-maze test (Fig. 7B). Overall, directly supplementing BiP levels in the hippocampus is sufficient to improve learning in the APPNL-G-F KI mouse model of AD.
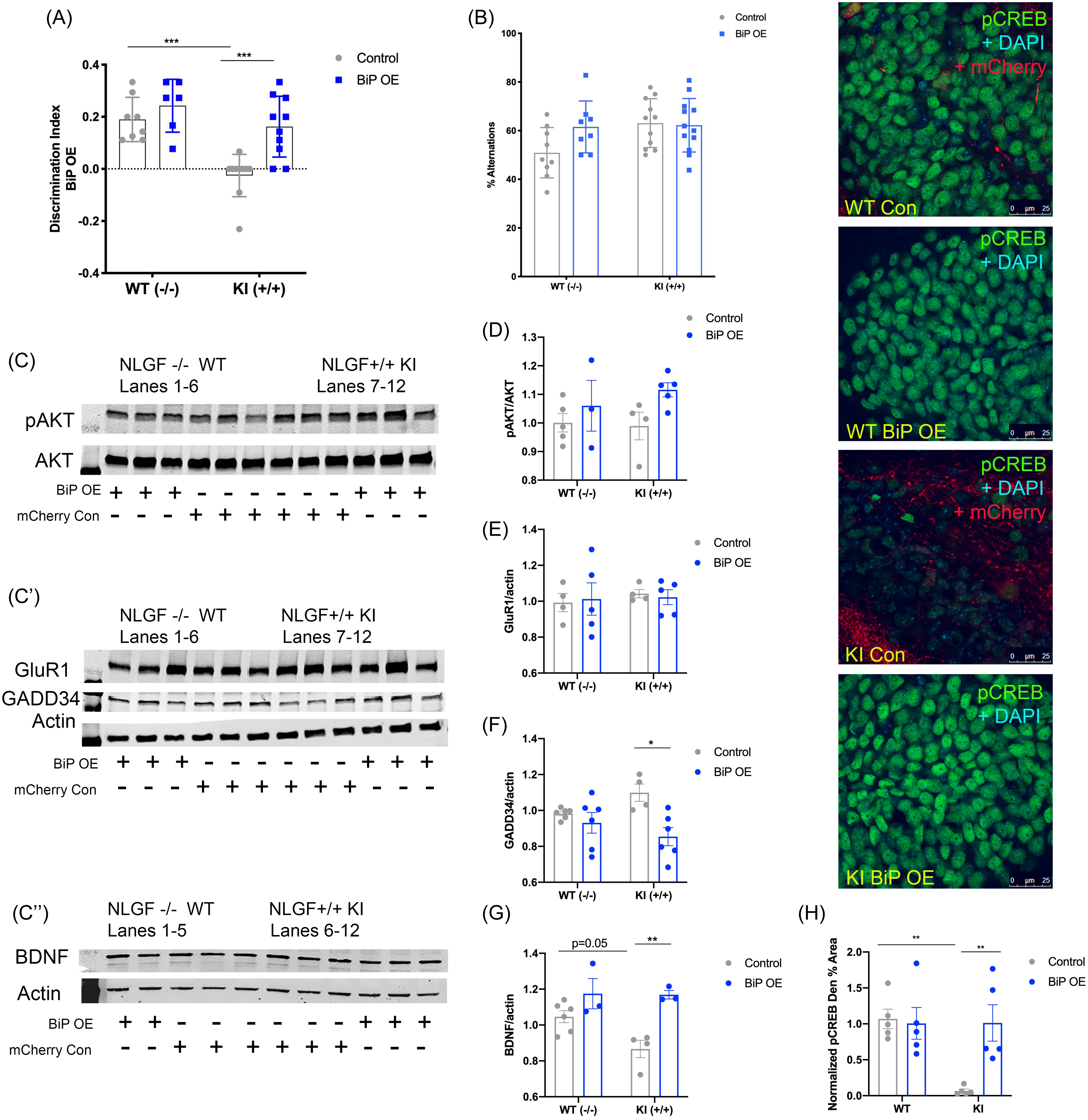
(A) Discrimination index of the SOR test. (B) Percent alternations in the Y-maze test. (C–C″) Representative western-blot images; each panel is a different animal. Membranes are cut along the MW markers following blocking to use the two-channel Odyssey system and varying molecular weights for the proteins of interest to probe for multiple markers within one membrane: (C) pAKT and AKT, ∼65 kDa; (C′) GluR1 ∼110 kDa, GADD34 ∼100 kDa, Actin ∼42 kDa; and (C″) BDNF dimer ∼28 kDa, Actin ∼42 kDa. (D) Quantification of western-blot analysis for pAKT/AKT. (E) Quantification of western-blot analysis for GADD34/actin. (F) Quantification of western-blot analysis for GluR1/actin. (G) Quantification of western-blot analysis for BDNF/actin. Right: Confocal images of the hippocampal dentate gyrus across groups. (H) Quantification of pCREB immunofluorescence normalized to APPWT WT mice, mean ± SE percent area within hippocampal dentate gyrus sections. n = 3–6 animals per group for western blot and n = 5 for immunohistochemical assays. (All data presented as mean ± SE; all quantifications analyzed via two-way ANOVA with Tukey post hoc correction for multiple comparisons, *p < 0.05, **p < 0.01, ***p < 0.001).
BiP overexpression induces CREB activation and BDNF levels in the APPNL-G-F KI mice
We wanted to examine a mechanism for BiP-overexpression-induced improvement in cognition. We probed for CREB phosphorylation in AAV-BiP and AAV-mCherry control-injected APPNL-G-F KI and APPWT WT mice. APPNL-G-F KI AAV-control-injected mice displayed significantly less p-CREB in the dentate gyrus compared to APPWT WT controls (p < 0.01; Fig. 7; Supplemental Table 2). With BiP overexpression injections, APPNL-G-F KI mice showed increased p-CREB compared to APPNL-G-F KI control-injected mice (p < 0.01; Fig. 7; Supplemental Table 2), suggesting that BiP overexpression in the hippocampus is sufficient to increase CREB phosphorylation. As in the other studies, we also examined p-AKT and GADD34 levels. We found that AKT phosphorylation did not significantly change across groups (Fig. 7C,D). In addition, APPNL-G-F KI control-injected mice did not display an increase in GADD34 compared to control-injected APPWT WT littermates (Fig. 7C′,F). However, BiP overexpression in the APPNL-G-F KI mice led to a reduction in GADD34 levels compared to control-injected APPNL-G-F KI mice (p < 0.05; Fig. 7C′,F). Furthermore, as with PBA and saline-treated mice, GluR1 levels did not change, regardless of genotype or injection (Fig. 7C′,E). However, APPNL-G-F KI control-injected mice exhibited less BDNF levels as compared to APPWT WT control-injected mice (p = 0.05; Fig. 7C″,G), and BiP overexpression led to an increase in BDNF in APPNL-G-F KI mice (p < 0.01; Fig. 7C″,G). Altogether, these results demonstrate that local hippocampal BiP overexpression is sufficient to improve learning in these APPNL-G-F KI mice through increasing CREB phosphorylation and BDNF.
Discussion
In this study, we postulated that using chaperone treatment in an APP KI mouse model of AD would improve proteostasis and cognition. Encouragingly, our results indicate that chaperone therapy is sufficient to reduce chronic UPR activity and improve cognitive performance in the SOR test in the APPNL-G-F mouse model of AD. In particular, the control-treated APPNL-G-F KI mice displayed reduced levels of XBP1s, regardless of age, which is restored with PBA treatment. Importantly, changes in XBP1s levels are linked to APP processing, as XBP1s is known to promote disintegrin and metalloproteinase 10 (ADAM10)10,40. ADAM10 is an alpha-secretase that cleaves APP in a non-amyloidogenic manner11. In AD, ADAM10 levels are reduced, and APP is preferentially cleaved by BACE1, a beta-secretase, and gamma-secretases, ultimately generating Aβ42 in the amyloidogenic pathway11,13. Under healthy conditions, APP has a functional role in the synapse, involved in cell adhesion and synapse stability12,62. We show here that ADAM10 levels are reduced in APPNL-G-F KI mice but are restored with PBA treatment, correlated with the changes we observed in XBP1s staining. Thus, modulating the UPR via chaperone therapy has a direct impact on APP processing.
While we did not observe any changes in the number of plaques with PBA treatment in the young mice, we did observe a significant reduction in plaque number in the 12–14-month-old APPNL-G-F mice. Consistent with the change in plaques, we observed a reduction in soluble Aβ42 with PBA treatment in these late-stage intervention APPNL-G-F KI mice. Soluble Aβ oligomers are known to lead to cognitive deficits even in the absence of plaques61, and there is literature showing that Aβ plaques are not the most toxic Aβ species60,61,63. Indeed, Aβ plaques have been observed in the brains of non-AD cognitively normal patients and animals, indicating that larger aggregates do not cause cognitive impairments and that Aβ oligomers are the more toxic species of Aβ64–68. Overall, our results are consistent with the changes observed in ADAM10, indicating that modulating the UPR can impact Aβ42 amounts. However, while ADAM10 levels were increased with chaperone treatment, ADAM10 itself has been associated with long-term depression (LTD) and synaptic downscaling69,70. Therefore, the changes we observed in ADAM10 could be linked to improved pathology but do not fully explain the improvement in cognition with chaperone treatment. It is likely that a combination of factors, including the increase in p-CREB and BDNF together with reduced Aβ42 as a result of increased ADAM10, contribute to the improved learning phenotype.
Supplementing chaperone levels to modulate the UPR affects other cellular processes, including those involved in memory formation. Promisingly, the impaired cognition observed in the APPNL-G-F KI saline-treated and AAV-control-injected mice was rescued by PBA and BiP, respectively. Importantly, our data indicate that PBA treatment, even administered at a late stage in disease progression, was sufficient to restore cognition in the APPNL-G-F KI mice. Our findings align with a recently published study that demonstrates the crucial role of ER genes, especially those of BiP, in long-term memory71. Similar to our observations, Chatterjee et al found that BiP overexpression rescued long-term memory deficits in a tau-based mouse model of ADRD. That study, along with the data presented here, highlights the significance of chaperone proteins in the consolidation of memory. While in the present study we used the SOR test, future experiments should conduct a repertoire of tasks, including fear conditioning or the Morris Water Maze test, to corroborate our findings.
We have previously shown that supplementing chaperone levels in aged mice reduces PERK activation, derepressing global protein translation, which is necessary for memory formation. In this aging model, we showed that chaperone treatment reduced PERK signaling and GADD34 levels, leading to an increase in CREB activation correlated with improved learning in the aged mice30. GADD34 upregulation prevents phosphorylation of AKT, a CREB kinase, thus impairing CREB phosphorylation and, thus, cognition50,51. In the present study, we examined if a similar mechanism was involved in this mouse model of AD. Overall, our results show similar changes in GADD34 and p-AKT with PBA treatment and BiP overexpression, suggesting that a mechanism comparable to what we previously identified in a model of aging could apply to this APPNL-G-F KI model of AD, indicating that some age-related cellular processes may be involved in AD progression. Furthermore, it is known that in AD there is a reduction of AMPA receptor levels that is linked to impaired cognition72,73. Here, we report no change in the GluR1 subunit of AMPA receptors via western blot using whole-hippocampal homogenate. However, this method lacks regional specificity, thus inhibiting the measurement of potential local hippocampal differences in AMPA levels. Many of the immunohistochemical assays revealed hippocampal region-specific changes, and thus it is possible that GluR1 levels also change in hippocampal subregions. A future direction for this work would be to examine synaptosomal preparations, which could inform more nuanced changes in AMPA distribution directly at the synapse.
Under normal conditions, the UPR functions as an adaptive response to acute ER stress19,21. In the short term, PERK leads to attenuated translation, while ATF6 and IRE1 lead to increased chaperone synthesis and activation of protein degradation pathways19,21. However, in the presence of unresolved ER stress, chronic UPR activity initiates pro-apoptotic signaling through the PERK-CHOP pathway21,74. Indeed, it has been shown that the pro-survival arms of the UPR, namely IRE1-induced XBP1 splicing that promotes chaperone synthesis, are attenuated during persistent ER stress74. This change is critical, as it is thought to serve as a turning point for the cell, from survival mechanisms to the initiation of apoptosis. In the case of neurodegenerative diseases, and AD specifically, proteostasis is chronically dysfunctional, likely contributing to the pathological aggregations observed and the progression of neurodegeneration6,16,27,28,35. Interestingly, we observed reduced levels of BiP and XBP1s in the APPNL-G-F KI mice, which were rescued with PBA treatment from weaning and late-stage intervention. This suggests that supplementing chaperone levels, even later in disease progression, promotes pro-survival UPR activation, as enhancing XBP1s levels could increase protein folding capacity by promoting endogenous BiP. Furthermore, late-stage intervention saline-treated APPNL-G-F KI mice displayed an increase in CHOP compared to age-matched APPWT WT saline-treated mice, suggesting that if left untreated, this APP KI mouse model of AD leads to UPR-related pro-apoptotic signaling. CHOP levels were reduced with PBA treatment, again providing evidence that supplementing chaperone levels promotes an adaptive UPR response and reduces chronic UPR signaling.
ER stress is believed to play an important role in AD. Other animal models of AD display ER stress; however, because these models overexpress membrane proteins like APP or PS1, ER stress may be induced nonspecifically as these proteins can be misfolded and aggregate in the ER75. Thus, the use of the APPNL-G-F KI model, which increases Aβ without overexpressing APP, provides a unique tool to discern if ER stress in these AD models is due to Aβ pathology as opposed to overexpressed APP. Interestingly, the research group that developed the APPNL-G-F KI model for AD also examined ER stress in these mice75. The authors claim that there is no induction of ER stress in these APPNL-G-F KI mice and thus that ER stress observed in other AD models is nonspecific75. However, those observations are limited by low animal numbers and western-blot assays using whole brain region homogenates, overlooking potential effects in subregions or specific neural populations. There is a robust volume of literature that has examined ER stress in mouse models and postmortem human tissue and has reported increased ER stress, including increased PERK activation6,76–78. It is therefore vitally important to distinguish effects that are due to specific disease pathology as opposed to changes resulting from the limitations inherent in the animal models used (i.e., APP overexpression). We observed increases in several markers of ER stress in the APPNL-G-F KI saline-treated and control-injected mice, which could be due in part to using higher animal numbers as well as IF to examine hippocampal subregions.
It is also important to note that PBA, in addition to acting as a protein chaperone, also functions as a histone deacetylase (HDAC) inhibitor79,80. In this study, we found that PBA treatment did not alter the acetylation of histones H3 and H4. Our data, which focus on the chaperone function of PBA, have been substantiated by Chatterjee et al71, who showed that sodium butyrate, which acts solely as an HDAC inhibitor, does not rescue cognition in an AD model. Further substantiating a role for chaperones, our data also indicate that local hippocampal BiP overexpression was sufficient to improve both memory and proteostasis in APPNL-G-F KI mice. These experiments corroborate the role of PBA as a chaperone in proteostasis and cognition and exemplify the role of ER stress in behavior.
Altogether, the results presented here are consistent with an extensive body of literature that has studied the role of UPR signaling in pathology and memory in AD6,27. PERK signaling is known to be increased in the tissue of AD patients as well as in various animal models of AD6,27. Critically, several studies have shown that reducing UPR activity or modulating the UPR improves memory in AD models44,45,81. It has been shown that inhibiting phosphorylation of eIF2a, downstream of PERK and responsible for attenuating global translation, improves memory and synaptic plasticity in models of AD44,82,83. Another study demonstrated that ATF4 is linked to impaired memory and synaptic plasticity84. Key to our discussion are the studies linking XBP1s to BDNF41,85. It has been shown that genetic depletion of XBP1s leads to deficits in synaptic plasticity and impaired learning, as well as reduced bdnf expression, while viral overexpression of XBP1s results in enhanced LTP, improved memory, and increased BDNF41,85. BDNF is a growth factor known to promote memory formation56–58, and its expression is impaired in AD, which is thought to be linked to the hallmark cognitive impairments86–88.
Thus, we propose that there are multiple pathways that are beneficially modulated by chaperone treatment, as illustrated in Figure 8. Supplementing chaperone levels by increasing XBP1s serves to ameliorate disease-related characteristics in several ways. The first is that XBP1s drives ADAM10 to reduce aberrant Aβ42. XBP1s also leads to improved memory formation via promoting BDNF, as well as improved proteostasis by promoting the synthesis of the chaperone BiP. Meanwhile, PERK signaling is reduced with both PBA treatment and local hippocampal BiP overexpression, suggesting that the protein translation block via peIF2α is lifted, allowing for normal translation to proceed. Phosphorylated CREB levels are also increased with chaperone treatment, which could in part be due to a reduction in PERK-ATF4-GADD34 signaling. In the late-stage intervention paradigm, PERK-CHOP signaling is ameliorated, suggesting that UPR-related pro-apoptotic signaling is reduced. It is these collective changes that shift the cell toward adaptive pro-survival signaling, making chaperone supplementation an appealing target for the treatment of AD.
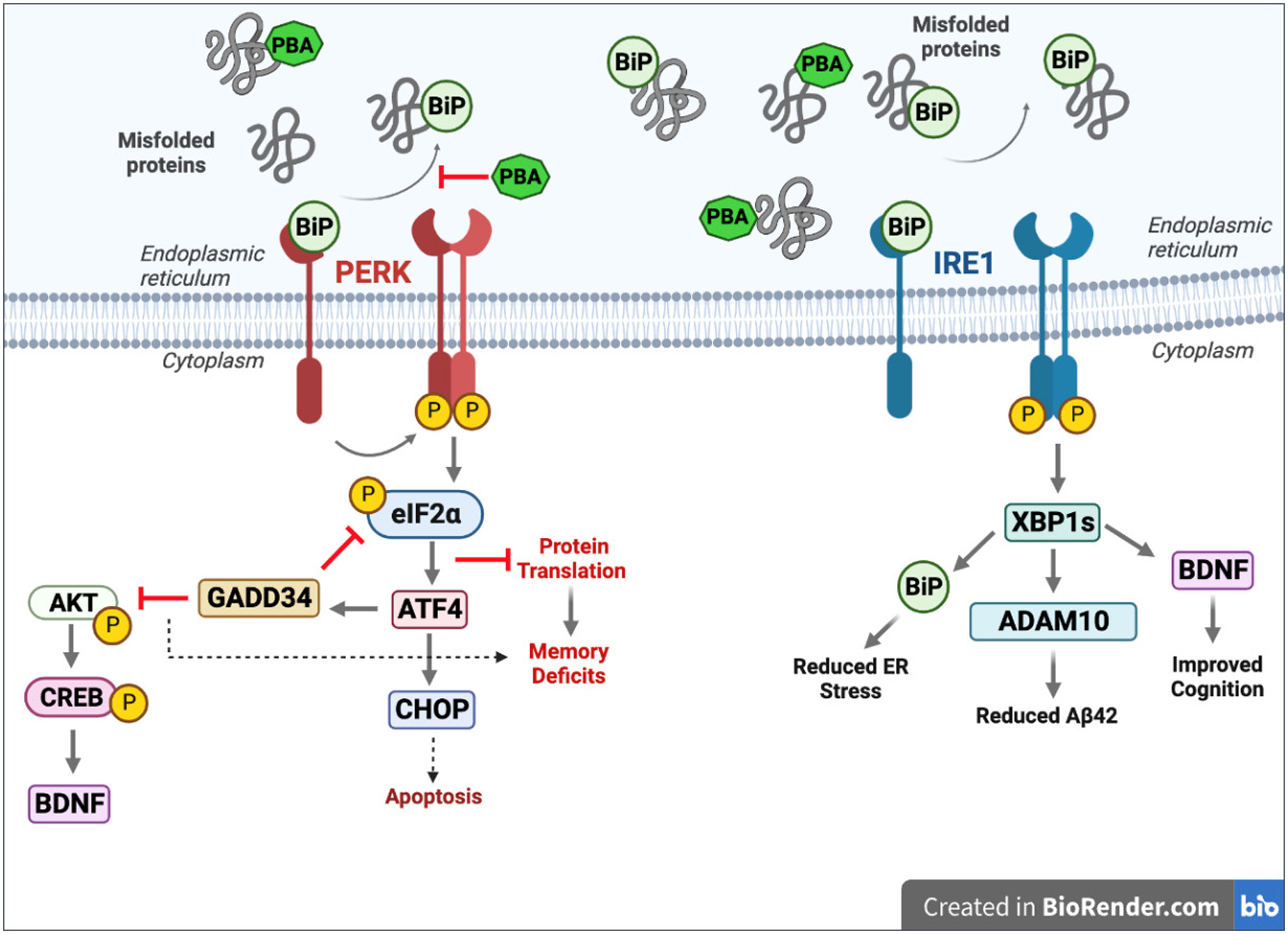
Perk activation following BiP dissociation leads to inhibited protein translation via p-eIF2a, increased GADD34, reduced p-CREB signaling, and increased CHOP. Chaperone therapy reduces PERK activation and promotes IRE1 activation, which leads to increased XBP1s, BiP, ADAM10, and BDNF.
Conclusion
Our results show that supplementing protein chaperone levels improves proteostasis and cognition in this APPNL-G-F KI mouse model of AD. Chaperone treatment leads to an adaptive UPR response that is linked to non-amyloidogenic APP processing and increased CREB phosphorylation. Encouragingly, even late-stage chaperone treatment improves cognition and proteostasis. Collectively, this work could provide valuable insight into the development of novel therapeutics for this debilitating and pervasive disease.
Acknowledgments
The authors wish to acknowledge and thank Robert Komlo for assistance with weekly intraperitoneal injections and Brendan Keenan of the Sleep Statistical Core for assistance with statistical analyses.
Funding
R01 AG064231 Cellular and Molecular Basis of Sleep Loss Neural Injury in Alzheimer Disease R56 AG061057 Pancreatic proteostasis connects sleep disruption to Alzheimer’s Disease.
Author Contributions
J.M.H. designed and conducted the experiments, analyzed the data, and wrote this article. E.S. conducted western-blot experiments. K.S. conducted immunofluorescence (IF) and plaque counts. N.N. conceptualized and designed the study, provided the resources, interpreted data, edited, and revised this article.
Competing Interests
The authors have no conflicts of interest to declare.
Data and Materials Availability
The raw data supporting the conclusions in this article will be made available upon request.
Supplementary Materials
Supplemental information can be found here Supplementary.